

Next Generation Sequencing in Autism Spectrum Disorder
Jiani Yin 1,2 ![]() , David Oleson 3
, David Oleson 3![]() , Christian P. Schaaf 1,2,*
, Christian P. Schaaf 1,2,*![]()
- Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA
- Jan and Dan Duncan Neurological Research Institute at Texas Children's Hospital, Houston, TX, USA
- Baylor College of Medicine, Houston, TX, USA
* Correspondence: Christian P. Schaaf ![]()
Received: August 31, 2017 | Accepted: February 09, 2018 | Published: February 24, 2018
OBM Genetics 2018, Volume 2, Issue 1 doi:10.21926/obm.genet.1801014
Academic Editors: Ute Moog and Domenico Coviello
Special Issue: Next Generation Sequencing
Recommended citation: Yin JN, Oleson D, Schaaf CP. Next Generation Sequencing in Autism Spectrum Disorder. OBM Genetics 2018;2(1):014; doi:10.21926/obm.genet.1801014.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Autism spectrum disorder is a clinically heterogeneous condition, characterized by social deficits, language impairment, repetitive behaviors, and restricted interest. Autism displays significant genetic heterogeneity. In the past one and a half decades, next generation sequencing has enabled identification of many variants that predispose to autism. These discoveries have improved understanding of the disease etiology of autism spectrum disorder. In this review article, we will address how development of next generation sequencing has helped answer the following questions: 1. What are the modes of transmission/inheritance of autism? 2. What is the nature of genetic risk factors that contribute to autism? 3. Why is there a higher prevalence of autism in males than females?
Keywords
Next generation sequencing; autism spectrum disorder; genetic variants; chromosome conformation; male bias; machine learning
1. Introduction
The first cases of autism spectrum disorder (ASD) were reported by Leo Kanner in 1943, characterized by social disconnection and impaired language skills beginning from early childhood. In subsequent years, more individuals with autism were identified, with the number of males being about four times that of females [1]. In the late 1970s, twin studies estimated the heritability of autism spectrum disorder to be greater than 80% [2]. From these early encounters, several questions arose that have puzzled us for decades: 1. What are the modes of transmission/inheritance of autism? 2. What is the nature of genetic risk factors that contribute to autism? 3. Why is there a male bias in autism?
During early years, genetic linkage analysis and cytogenetic tools were leveraged to identify genes involved in the pathophysiology of autism. A few autism risk loci were initially identified, namely regions 2q, 7q31-q33, 15q11-q13, 16p, 19p, and Xp [3,4,5]. While they struggled with inconsistent genetic association findings [6,7,8,9] and low prevalence of mutations in autism individuals [10], scientists quickly recognized the huge genetic heterogeneity of autism [11]. The power of genetic loci detection at the time was limited by both the capacity to manipulate large sample sizes and resolution of detection tools. With the advent of next generation sequencing in the 2000s, we witnessed a blossom of gene discovery, achieved unprecedented resolution to the level of the single nucleotide, and revolutionized our understanding of the etiology of autism. In this review article, we will present a timeline for the identification of autism risk loci (Figure 1), discuss how next generation sequencing helped us to approach the previously raised questions, and present challenges and future directions. Since many review articles have summarized earlier findings, we will mainly focus on findings from the past five years.
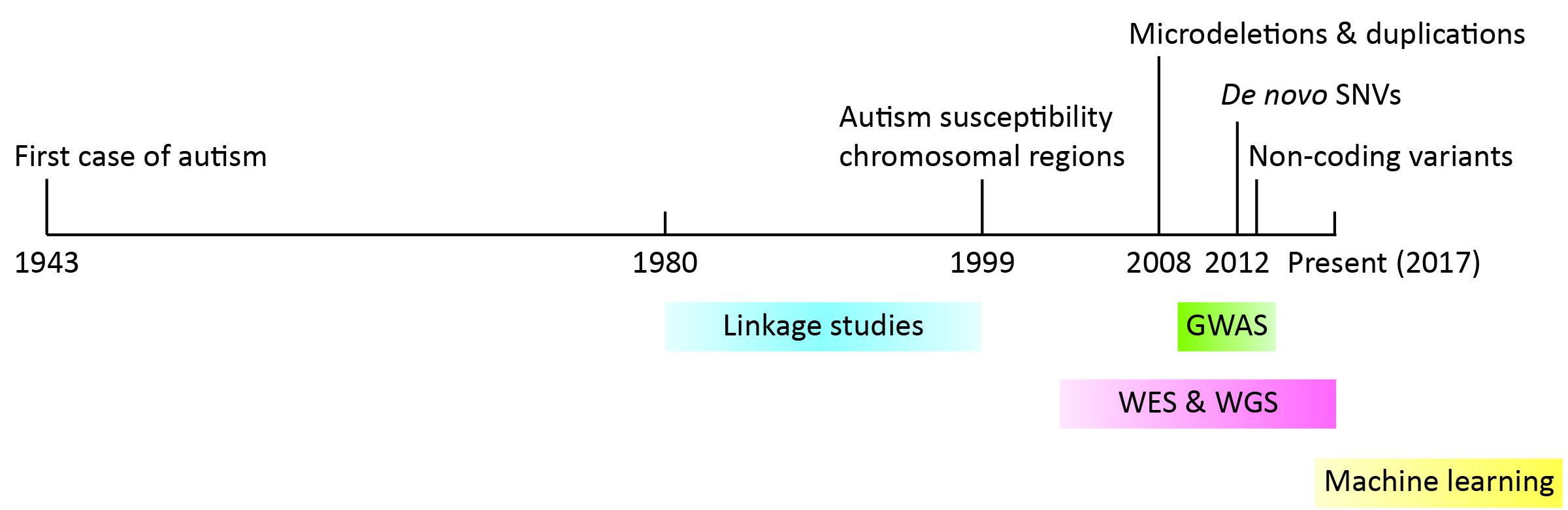
Figure 1 Timeline of important findings and methods used in the discovery of autism risk loci. Years are denoted on the black line. Findings are denoted at the top while methods are indicated at the bottom.
2. From Linkage Disequilibrium Analysis to Genome Wide Association Studies
In the 1980s and 1990s, linkage studies were used to tackle the mode of inheritance in autism, using familial cases of autism, and with most results coming out consistent with an autosomal recessive inheritance model [12]. Human leukocyte antigen (HLA) genes and a few neuronal genes were tested as candidates. The genes tested included the HLA loci [13], fragile-X syndrome gene FMR1 [14], serotonin transporter gene HTT [7,9], GABA receptor subunit gene GABRB3 [9,15], among others. However, association of these genes with ASD was either insignificant or inconsistent among different studies. Genome-wide screens with genetic markers, typically using a collection of 50-100 autism families, identified several autism susceptibility regions [3,4,16,17], scattered across different chromosomes. Direct sequencing of genes that had been reported in autism probands in follow-up studies using larger cohorts usually led to conclusions that mutations in these genes were uncommon to autism [10]. No study was able to narrow the candidate regions to single genes, except for those known to cause syndromic autism. In the late 1990s, a consensus was reached that a fairly large number of genes may be involved in ASD, and that each individual gene may have a small effect [11].
With the implementation of next generation sequencing and accomplishment of the human genome project, scientists gained access to a larger number of tightly-spaced genetic markers, or single nucleotide polymorphisms (SNPs). The hope was to identify common variants underlying common disease, such as ASD, through regional or genome-wide association studies (GWAS) with collection of 1000-3000 patients and an unaffected control group. A handful of loci were reported as plausible risk loci for ASD [18,19,20,21], however, only SNPs near CDH10 and CDH9 and SNPs at 1p13.2 region were reproduced using independent cohorts [22,23,24]. Other studies found no association signal that met statistical significance [25,26,27]. Stratifying the samples by sex or subphenotypes helped to identify new candidate loci [28,29], albeit this strategy met the same bottleneck of reproducibility. These findings highlighted the genetic heterogeneity of autism, and implicated that ASD may be largely attributed to rare variants.
3. Impact of Single Nucleotide Variants in Genes
With advancement of next generation sequencing technology, we were eventually capable of reading the genome at single nucleotide resolution. Studying families with shared ancestry greatly enhanced identification of risk loci with a recessive mode of inheritance [30,31,32], which included UBE3B, CLTCL1, NCKAP5L, ZNF18, PCDH10, DIA1, NHE9, AMT, PEX7, SYNE1, and others.
In 2012, three articles published back-to-back in Nature, highlighted the impact of de novo single nucleotide variants (SNVs) [33,34,35] on autism spectrum disorder. They performed whole exome sequencing (WES) on 500-1000 individuals respectively, and reported a significantly increased rate of gene-disrupting or loss-of-function de novo SNVs in subjects with ASD. The prevalence of these de novo changes was positively correlated with paternal age. Several autism risk genes with dominant effects were identified by these and other groups, including SCN1A, SCN2A, CHD2, CHD8, KATNAL2, NTNG1, GRIN2B, LAMC3, DYRK1A, DAT1, SHANK1, SHANK3, SYNGAP1, TRIP12, PAX5, TCF20 [33,34,35,36,37,38,39,40,41]. Many of these genes are involved in FMRP-associated pathways or in the β-catenin/chromatin-remodeling protein network [34,42]. Taken together, de novo SNVs may account for around 5-20% of ASD cases [43].
SFARI database is an involving database for genes implicated in autism susceptibility. We carefully curated all the SFARI genes associated with autism for their modes of inheritance, and found 432 dominant genes, 48 recessive ones, and 44 X-linked, with 357 genes showing insufficient evidence for a disease-driving effect (Figure 2, Supplementary Table 1). Beyond what was mentioned above, somatic mutations [44,45], human-specific regions and developmental programs [46,47], and increased burden of deleterious mutations in essential genes [48] have also been implicated in ASD. Clinically and functionally validated autism risk genes have been reviewed in the following reference [49].
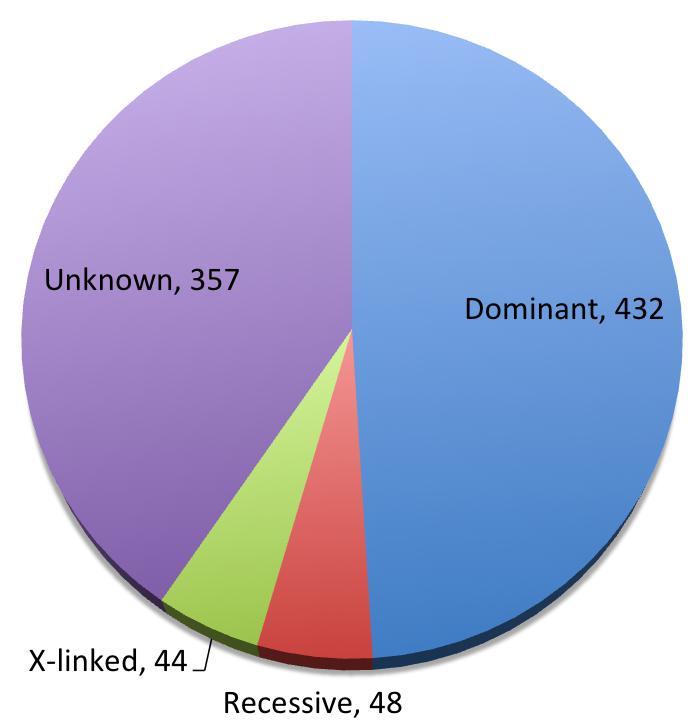
Figure 2 Pie chart of inheritance patterns of SFARI genes.
4. New Cytogenetic Findings
Cytogenetic analysis represents another important means of uncovering the genetic cause of ASD. Next generation sequencing and the human genome project provided a reference genome for implementation of comparative genomic hybridization microarray (aCGH), which has a much higher resolution than traditional techniques. Such improvements led to identification of many inherited copy number variations (CNVs) that are associated with autism, including 1p34.2-p34.3, 2q23.1, 3q29, 7q11.23, 15q13.3, 16p11.2, 16q24.3, and 17q12-13 deletions or duplications [50]. Similarly, de novo CNVs present substantial risk for ASD, explaining approximately 2-7% of idiopathic autism cases [51,52,53,54,55,56]. Exonic CNVs and whole gene deletions or duplications that were present only in affected individuals overlapped with known ASD candidate genes and identified previously unreported loci [57,58]. Notably, in the majority of autism families that harbor inherited or de novo CNVs, not all affected individuals carry the mutation, indicating that there are other risk factors that contribute to the disease manifestation [55].
Balanced chromosomal abnormalities (BCA) are powerful in pinpointing specific genes as well, in that the breakpoints usually disrupt expression of one to two genes. Next generation sequencing largely improved the resolution of breakpoint localization, and several potential candidate genes were implicated by de novo translocations and inversions, such as TRIP8 [59], REEP3 [59], NRXN1 [60], CSMD3 [61], and RAB11FIP5 [62]. Association of these genes with ASD will be better supported by additional evidence of variants within the genes and functional studies.
Compared with aCGH, whole genome sequencing (WGS) provides more detailed information about chromosomal rearrangements, albeit it has proven quite challenging to obtain copy number information. Recent progress involves identification of novel autism risk loci [63] and discovery of complex structural variations, in which different types of chromosomal rearrangements were generated at a single loci [64]. Application of de novo assembly of sequenced genomes uncovered previously undetectable mutations [65]. However the “gap” between read lengths of NGS (100 bp) and resolution of aCGH (5 kb) needs to be covered and will likely reveal many more variants.
To have a better idea of which autism risk loci were subject to copy number variation versus single nucleotide change, we curated all SFARI genes for evidence of SNV, CNV, and BCA. Our results show a substantial overlap between genes affected by SNV and CNV (Figure 3, Supplementary Table 1). In the future, it will be interesting to identify SNVs in genes that were initially associated with in CNVs alone.
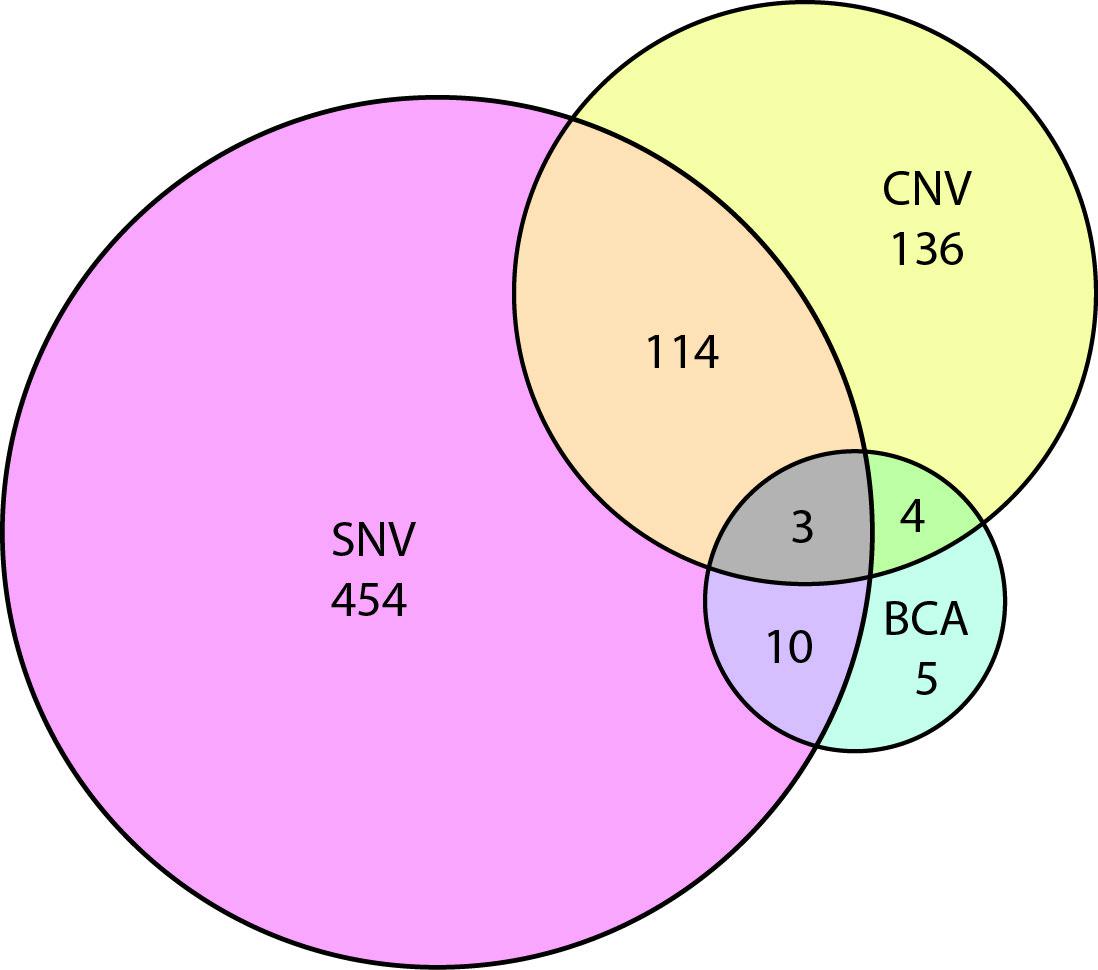
Figure 3 Venn diagram showing the overlap of autism risk genes with evidence of copy number variation (CNV), single nucleotide variation (SNV), or balanced chromosomal abnormality (BCA).
5. Non-coding Variants and Chromosome Conformation
CNVs and SNVs that affect specific genes have been established as risk factors for autism spectrum disorder, which in total account for approximately 20% of ASD individuals [66]. Little effort had been invested into the vast intergenic regions of the genome until 2013, when rare inherited CNVs affecting only intergenic regions were reported to affect 3% of ASD cases [66]. Subsequently whole genome sequencing on ASD families, in which no CNV or SNV had been implicated, showed that probands had a significant enrichment of de novo loss-of-function mutations in DNase1 hypersensitive sites, which are putative regulatory regions [67]. MicroRNAs, long non-coding RNAs, and regions close to splicing sites have all been implicated in association with ASD through WGS and RNA profiling [68,69,70,71]. Dysregulation of the epigenome and chromosome conformation may complement DNA mutations in the pathogenesis of autism [72,73,74], studies of which were enabled by ChIP-seq (Chromatin immunoprecipitation sequencing) and other techniques.
6. Rethinking Common Variants
Notably, some of the regulatory regions are enriched for genetic variants from GWAS studies [75]. Furthermore, 3D chromatin interaction map in the developing human brain showed that some GWAS loci regulate putative neuropsychiatric disease gene expression [74]. These findings urge us to rethink about common variants and their contribution to ASD and other neuropsychiatric disorders. In fact, some studies found that combinatorial effects of hypomorphic or common variants account for a major part of ASD heritability, yet rare variation contributes to variance in liability [76,77].
Years of experience have taught us that such common variations scatter across the entire genome, with an impressive total number and a small individual impact. Machine learning emerged as a potential means of detecting the pattern of variation, dissecting genetic components and subphenotypes, as well as improving clinical diagnosis. Pioneering studies have applied machine learning to develop an observation-based diagnostic classifier of autism [78], and to predict new candidate genes based on features of the known ones [79]. We foresee unprecedented findings from machine learning of patient and unaffected individuals at a multi-dimensional level.
7. Possible Explanations for Male Bias
Autism has been consistently reported to affect more males than females, with the commonly referenced male-to-female ratio being 4:1. This cannot be explained simply by X-linked autism, due to the fact that there are more autosomal genes associated with autism than genes on the sex chromosome, and the prevalence of the X-linked genes alone does not account for the substantial difference between male and female cases. A female protective model has been proposed and supported by higher mutational load of de novo CNVs, de novo SNVs, and inherited variants in affected females compared to males [1,49,80]. Sex chromosomal genes, other sex related risk loci, hormone factors, and innate sex differences in neurodevelopmental and immune systems that may contribute to the sex bias of ASD have been identified [1,81,82]. While they cannot fully explain the sex-skewed prevalence of autism, one of the most convincing explanations emerged from an RNA-seq study, which discovered that genes highly expressed in males were significantly enriched for those that were upregulated in postmortem ASD brains [83]. Further investigations into naturally occurring sexual dimorphism and gene-environment interactions will help elucidate the mechanism of male bias in autism.
8. Overlap with Other Neuropsychiatric Disorders
Individuals with ASD show high rates of comorbidity with other neuropsychiatric disorders, including intellectual disability (ID, 45%), attention deficit hyperactivity disorder (ADHD, 28-44%), clinical depression (12-70%), epilepsy (8-30%), schizophrenia (SCZ), and bipolar disorder [84]. A number of CNVs and specific genes are associated with multiple neuropsychiatric conditions [85,86], suggesting genetic correlation between these different neuropsychiatric disorders. Identification of specific risk loci allowed us to further investigate what genetic factors may be shared or distinct. Studies demonstrate that few genes are specific to ASD. Risk loci of autism and intellectual disability have extensive overlap [87], and schizophrenia risk genes overlap with genes implicated in autism and ID [88]. On the other hand, ADHD may be genetically different from autism [89,90].
9. Clinical Diagnosis
Diagnosis of autism spectrum disorder can be difficult, given the variation in clinical manifestations and the absence of medical tests or strong, measurable biomarkers. Currently, individuals with autism are diagnosed based on the DSM-5 criteria (American Psychiatric Association, 2013). With mounting knowledge of autism susceptibility loci, next generation sequencing has great potential to facilitate clinical diagnosis of ASD, to refine clinical features of related syndromes, and may suggest specific interventions to modify phenotype, the power of which has been demonstrated by a few groups [91,92]. However, autism is and will continue to be a clinical diagnosis. No single genetic test will make a diagnosis of autism, in part because of incomplete penetrance, pleiotropy, and variable expressivity.
10. Challenges and Future Directions
With an overwhelming, accelerating number of new variants being identified in individuals with autism, one of the challenges is to prevent false-positive reports of causality. A guideline has been put forward to regulate the process from study design to both gene-level and variant-level assessments of evidence [93]. During variant interpretation, it is always helpful to refer to the allele frequency in the ExAC database, which contains exomes from approximately 60,000 individuals, who had no clinical manifestations and various ethnic backgrounds (exac.broadinstitute.org). There also exist databases that can help determine whether a specific gene/variant is linked to ASD, for example the SFARI gene (https://sfari.org/resources/sfari-gene), the Geisinger Developmental Brain Disorder Genes database (http://geisingeradmi.org/care-innovation/studies/dbd-genes/), and ClinGen (https://www.clinicalgenome.org).
Pipelines for analysis of variants in gene-coding regions have been maturing in the past decade. However, non-coding variants were commonly filtered out, with a lack of appreciation of their potential consequence. Development in computational algorithms and functional assays for these non-coding regions promise to open new avenues of understanding the genetics of autism.
11. Conclusions
We started this review by asking the following questions: 1. What are the modes of transmission/inheritance of autism? 2. What is the nature of genetic risk factors that contribute to autism? 3. Why is there a male bias in autism? Obviously, there are no easy answers to these questions. Copy number variations, X-linked inheritance, autosomal recessive or dominant inheritance, de novo mutations, and common variants, all play a role in autism susceptibility. There is tremendous genetic heterogeneity in autism with several hundred genes contributing to the overall prevalence. Many of the well-established genes and proteins are involved in synaptic and chromatin remodeling pathways. Sexual dimorphism of gene expression contributes to the female protective effect of autism. Other innate differences in neurodevelopmental and immune systems are waiting to be uncovered. With more large-scale sequencing projects, unprecedented advances in single-cell sequencing, chromosome conformation capture, and human in vitro models, we foresee a better understanding of the etiology of autism at multiple levels.
Acknowledgments
Dr. Christian Schaaf is the Joan and Stanford Alexander Chair for Neuropsychiatric Genetics at Texas Children’s Hospital.
Author Contributions
Jiani Yin and Dr. Christian Schaaf conceived the outline of the manuscript. Jiani Yin and David Oleoson wrote the manuscript. Dr. Christian Schaaf supervised the work.
Funding Source
None.
Competing Interests
The authors have declared that no competing interests exist.
Additional Materials
The following additional materials are uploaded at the page of this paper.
References
- Werling DM, Geschwind DH. Sex differences in autism spectrum disorders. Curr Opin Neurol. 2013;26:146-153. [CrossRef] [Google scholar] [PubMed]
- Folstein S, Rutter M. Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psyc. 1977;18:297-321. [CrossRef] [Google scholar] [PubMed]
- Philippe A, Martinez M, Guilloud-Bataille M, Gillberg C, Råstam M, Sponheim E, et al. Genome-wide scan for autism susceptibility genes. Hum Mol Genet. 1999;8:805-812. [CrossRef] [Google scholar] [PubMed]
- Barrett S, Beck JC, Bernier R, Bisson E, Braun TA, Casavant TL, et al. An autosomal genomic screen for autism. Collaborative linkage study of autism. Am J Med Genet. 1999;88:609-615. [CrossRef] [Google scholar]
- Thomas NS, Sharp AJ, Browne CE, Skuse D, Hardie C, Dennis NR. Xp deletions associated with autism in three females. Hum Genet. 1999;104:43-48. [CrossRef] [Google scholar] [PubMed]
- Salmon B, Hallmayer J, Rogers T, Kalaydjieva L, Petersen PB, Nicholas P, et al. Absence of linkage and linkage disequilibrium to chromosome 15q11‐q13 markers in 139 multiplex families with autism. Am J Med Genet. 1999;88:551-556. [CrossRef] [Google scholar]
- Jr EHC, Courchesne R, Lord C, Cox NJ, Yan S, Lincoln A, et al. Evidence of linkage between the serotonin transporter and autistic disorder. Mol Psychiat. 1997;2:247-250. [CrossRef] [Google scholar] [PubMed]
- Kluck SM, Poustka F, Benner A, Lesch K-P, Poustka A. Serotonin transporter (5-HTT) gene variants associated with autism? Hum Mol Genet. 1997;6:2233-2238. [CrossRef] [Google scholar] [PubMed]
- Maestrini E, Lai C, Marlow A, Matthews N, Wallace S, Bailey A, et al. Serotonin transporter (5-HTT) and-aminobutyric acid receptor subunit 3 (GABRB3) gene polymorphisms are not associated with autism in the IMGSA families. Am J Med Genet. 1999;88:492-496. [CrossRef] [Google scholar]
- Fon EA, Sarrazin J, Meunier C, Alarcia J, Shevell MI, Philippe A, et al. Adenylosuccinate lyase (ADSL) and infantile autism: absence of previously reported point mutation. Am J Med Genet A. 1995;60:554-557. [CrossRef] [Google scholar] [PubMed]
- Folstein SE, Bisson E, Santangelo SL, Piven J. Finding specific genes that cause autism: a combination of approaches will be needed to maximize power. J Autism Dev Disord. 1998;28:439-445. [CrossRef] [Google scholar] [PubMed]
- Mason-Brothers A, Mo A, Marazita ML. Evidence for autosomal recessive inheritance in 46 families with multiple incidences of autism. Am J Psychiat. 1985;142:187-192. [CrossRef] [Google scholar] [PubMed]
- Anne Spence M, Ritvo ER, Marazita ML, Funderburk SJ, Sparkes RS, Freeman BJ. Gene mapping studies with the syndrome of autism. Behav Genet. 1985;15:1-13. [CrossRef] [Google scholar] [PubMed]
- allmayer J, Pintado E, Lotspeich L, Spiker D, McMahon W, Petersen PB, et al. Molecular analysis and test of linkage between the FMR-1 gene and infantile autism in multiplex families. Am J Hum Genet. 1994;55:951-959. [Google scholar]
- Martin ER, Menold MM, Wolpert CM, Bass MP, Donnelly SL, Ravan SA, et al. Analysis of linkage disequilibrium in γ‐aminobutyric acid receptor subunit genes in autistic disorder. Am J Med Genet. 2000;96:43-48. [CrossRef] [Google scholar]
- Auranen M, Nieminen T, Majuri S, Vanhala R, Peltonen L, Järvelä I. Analysis of autism susceptibility gene loci on chromosomes 1p, 4p, 6q, 7q, 13q, 15q, 16p, 17q, 19q and 22q in Finnish multiplex families. Mol Psychiat. 2000;5:320. [CrossRef] [Google scholar] [PubMed]
- Ma DQ, Cuccaro ML, Jaworski JM, Haynes CS, Stephan DA, Parod J, et al. Dissecting the locus heterogeneity of autism: significant linkage to chromosome 12q14. Mol Psychiat. 2007;12:376-384. [CrossRef] [Google scholar] [PubMed]
- Chung R-H, Ma D, Wang K, Hedges DJ, Jaworski JM, Gilbert JR, et al. An X chromosome-wide association study in autism families identifies TBL1X as a novel autism spectrum disorder candidate gene in males. Mol Autism. 2011;2:18. [CrossRef] [Google scholar] [PubMed]
- de Krom M, Staal WG, Ophoff RA, Hendriks J, Buitelaar J, Franke B, et al. A common variant in DRD3 receptor is associated with autism spectrum disorder. Biol Psychiat. 2009;65:625-630. [CrossRef] [Google scholar] [PubMed]
- Weiss LA, Arking DE, Daly MJ, Chakravarti A, Brune CW, West K, et al. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802-808. [CrossRef] [Google scholar] [PubMed]
- Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet. 2008;82:160-164. [CrossRef] [Google scholar] [PubMed]
- Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, et al. Common genetic variants on 5p14. 1 associate with autism spectrum disorders. Nature. 2009;459:528-533. [CrossRef] [Google scholar] [PubMed]
- Torrico B, Chiocchetti AG, Bacchelli E, Trabetti E, Hervás A, Franke B, et al. Lack of replication of previous autism spectrum disorder GWAS hits in European populations. Autism Res. 2017;10:202-211. [CrossRef] [Google scholar] [PubMed]
- Xia K, Guo H, Hu Z, Xun G, Zuo L, Peng Y, et al. Common genetic variants on 1p13. 2 associate with risk of autism. Mol Psychiat. 2014;19:1212-1219. [CrossRef] [Google scholar] [PubMed]
- Anney R, Klei L, Pinto D, Regan R, Conroy J, Magalhaes TR, et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 2010:ddq307. [CrossRef] [Google scholar] [PubMed]
- Anney R, Klei L, Pinto D, Almeida J, Bacchelli E, Baird G, et al. Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum Mol Genet. 2012;21:4781-4792. [CrossRef] [Google scholar] [PubMed]
- Bonora E, Bacchelli E, Levy ER, Blasi F, Marlow A, Monaco AP, et al. Mutation screening and imprinting analysis of four candidate genes for autism in the 7q32 region. Mol Psychiat. 2002;7:289. [CrossRef] [Google scholar] [PubMed]
- Lu AT, Cantor RM. Allowing for sex differences increases power in a GWAS of multiplex Autism families. Mol Psychiatr. 2012;17:215-222. [CrossRef] [Google scholar] [PubMed]
- Liu X-Q, Paterson AD, Szatmari P, Autism Genome Project C. Genome-wide linkage analyses of quantitative and categorical autism subphenotypes. Biol Psychiat. 2008;64:561-570. [CrossRef] [Google scholar] [PubMed]
- Morrow EM, Yoo S-Y, Flavell SW, Kim T-K, Lin Y, Hill RS, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science (New York, NY). 2008;321:218-223. [CrossRef] [Google scholar] [PubMed]
- Chahrour MH, Timothy WY, Lim ET, Ataman B, Coulter ME, Hill RS, et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012;8:e1002635. [CrossRef] [Google scholar] [PubMed]
- Timothy WY, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259-273. [CrossRef] [Google scholar] [PubMed]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237-241. [CrossRef] [Google scholar] [PubMed]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246-250. [CrossRef] [Google scholar] [PubMed]
- Neale BM, Kou Y, Liu L, Ma’Ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242-245. [CrossRef] [Google scholar] [PubMed]
- van Bon BWM, Coe BP, Bernier R, Green C, Gerdts J, Witherspoon K, et al. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol Psychiat. 2016;21:126-132. [CrossRef] [Google scholar] [PubMed]
- Hamilton PJ, Campbell NG, Sharma S, Erreger K, Hansen FH, Saunders C, et al. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol Psychiat. 2013;18:1315-1323. [CrossRef] [Google scholar] [PubMed]
- Nemirovsky SI, Córdoba M, Zaiat JJ, Completa SP, Vega PA, González-Morón D, et al. Whole genome sequencing reveals a de novo SHANK3 mutation in familial autism spectrum disorder. PloS one. 2015;10:e0116358. [CrossRef] [Google scholar] [PubMed]
- O'Roak BJ, Stessman HA, Boyle EA, Witherspoon KT, Martin B, Lee C, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nature Commun. 2014;5:5595. [CrossRef] [Google scholar] [PubMed]
- Babbs C, Lloyd D, Pagnamenta AT, Twigg SRF, Green J, McGowan SJ, et al. De novo and rare inherited mutations implicate the transcriptional coregulator TCF20/SPBP in autism spectrum disorder. J Med Genet. 2014;51:737-747. [CrossRef] [Google scholar] [PubMed]
- Sato D, Lionel AC, Leblond CS, Prasad A, Pinto D, Walker S, et al. SHANK1 deletions in males with autism spectrum disorder. Am J Hum Genet. 2012;90:879-887. [CrossRef] [Google scholar] [PubMed]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285-299. [CrossRef] [Google scholar] [PubMed]
- O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585-589. [CrossRef] [Google scholar] [PubMed]
- D’Gama AM, Pochareddy S, Li M, Jamuar SS, Reiff RE, Lam A-TN, et al. Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron. 2015;88:910-917. [CrossRef] [Google scholar] [PubMed]
- Freed D, Pevsner J. The contribution of mosaic variants to autism spectrum disorder. PLoS Genet. 2016;12:e1006245. [CrossRef] [Google scholar] [PubMed]
- Doan RN, Bae B-I, Cubelos B, Chang C, Hossain AA, Al-Saad S, et al. Mutations in human accelerated regions disrupt cognition and social behavior. Cell. 2016;167:341-354. [CrossRef] [Google scholar] [PubMed]
- Liu X, Han D, Somel M, Jiang X, Hu H, Guijarro P, et al. Disruption of an evolutionarily novel synaptic expression pattern in autism. PLoS Biol. 2016;14:e1002558. [CrossRef] [Google scholar] [PubMed]
- Ji X, Kember RL, Brown CD, Bućan M. Increased burden of deleterious variants in essential genes in autism spectrum disorder. P Natl Acad Sci. 2016:201613195. [Google scholar]
- Yin J, Schaaf CP. Autism genetics–an overview. Prenatal diagnosis. 2016. [Google scholar]
- Nevado J, Mergener R, Palomares-Bralo M, Souza KR, Vallespín E, Mena R, et al. New microdeletion and microduplication syndromes: a comprehensive review. Genet Mol Biol. 2014;37:210-219. [CrossRef] [Google scholar] [PubMed]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science (New York, NY). 2007;316:445-449. [CrossRef] [Google scholar] [PubMed]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11. 23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863-885. [CrossRef] [Google scholar] [PubMed]
- Girirajan S, Brkanac Z, Coe BP, Baker C, Vives L, Vu TH, et al. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 2011;7:e1002334. [CrossRef] [Google scholar] [PubMed]
- Luo R, Sanders SJ, Tian Y, Voineagu I, Huang N, Chu SH, et al. Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am J Hum Genet. 2012;91:38-55. [CrossRef] [Google scholar] [PubMed]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87:1215-1233. [CrossRef] [Google scholar] [PubMed]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477-488. [CrossRef] [Google scholar] [PubMed]
- Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009;5:e1000536. [CrossRef] [Google scholar] [PubMed]
- Celestino-Soper PBS, Shaw CA, Sanders SJ, Li J, Murtha MT, Ercan-Sencicek AG, et al. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum Mol Genet. 2011;20:4360-4370. [CrossRef] [Google scholar] [PubMed]
- Castermans D, Vermeesch JR, Fryns J-P, Steyaert JG, Van de Ven WJM, Creemers JWM, et al. Identification and characterization of the TRIP8 and REEP3 genes on chromosome 10q21. 3 as novel candidate genes for autism. Eur J Hum Genet. 2007;15:422-431. [CrossRef] [Google scholar] [PubMed]
- Kim H-G, Kishikawa S, Higgins AW, Seong I-S, Donovan DJ, Shen Y, et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet. 2008;82:199-207. [CrossRef] [Google scholar] [PubMed]
- Floris C, Rassu S, Boccone L, Gasperini D, Cao A, Crisponi L. Two patients with balanced translocations and autistic disorder: CSMD3 as a candidate gene for autism found in their common 8q23 breakpoint area. Eur J Hum Genet. 2008;16:696-704. [CrossRef] [Google scholar] [PubMed]
- Roohi J, Tegay DH, Pomeroy JC, Burkett S, Stone G, Stanyon R, et al. A de novo apparently balanced translocation [46, XY, t (2; 9)(p13; p24)] interrupting RAB11FIP5 identifies a potential candidate gene for autism spectrum disorder. Am J Med Genet B. 2008;147:411-417. [CrossRef] [Google scholar] [PubMed]
- Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525-537. [CrossRef] [Google scholar] [PubMed]
- Collins RL, Brand H, Redin CE, Hanscom C, Antolik C, Stone MR, et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol. 2017;18:36. [CrossRef] [Google scholar] [PubMed]
- Brandler WM, Antaki D, Gujral M, Noor A, Rosanio G, Chapman TR, et al. Frequency and complexity of de novo structural mutation in autism. Am J Hum Genet. 2016;98:667-679. [CrossRef] [Google scholar] [PubMed]
- Walker S, Scherer SW. Identification of candidate intergenic risk loci in autism spectrum disorder. BMC Genomics. 2013;14:499. [CrossRef] [Google scholar] [PubMed]
- Turner TN, Hormozdiari F, Duyzend MH, McClymont SA, Hook PW, Iossifov I, et al. Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory DNA. Am J Hum Genet. 2016;98:58-74. [CrossRef] [Google scholar] [PubMed]
- Takata A, Ionita-Laza I, Gogos JA, Xu B, Karayiorgou M. De novo synonymous mutations in regulatory elements contribute to the genetic etiology of autism and schizophrenia. Neuron. 2016;89:940-947. [CrossRef] [Google scholar] [PubMed]
- Wu YE, Parikshak NN, Belgard TG, Geschwind DH. Genome-wide, integrative analysis implicates microRNA dysregulation in autism spectrum disorder. Nature neuroscience. 2016. [CrossRef] [Google scholar] [PubMed]
- Devanna P, Chen XS, Ho J, Gajewski D, Smith SD, Gialluisi A, et al. Next-gen sequencing identifies non-coding variation disrupting miRNA-binding sites in neurological disorders. Mol Psychiat. 2017. [CrossRef] [Google scholar] [PubMed]
- Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature. 2016;540:423-427. [CrossRef] [Google scholar] [PubMed]
- Berko ER, Suzuki M, Beren F, Lemetre C, Alaimo CM, Calder RB, et al. Mosaic epigenetic dysregulation of ectodermal cells in autism spectrum disorder. PLoS Genet. 2014;10:e1004402. [CrossRef] [Google scholar] [PubMed]
- Sun W, Poschmann J, del Rosario RC-H, Parikshak NN, Hajan HS, Kumar V, et al. Histone acetylome-wide association study of autism spectrum disorder. Cell. 2016;167:1385-1397. [CrossRef] [Google scholar] [PubMed]
- Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 2016;538:523-527. [CrossRef] [Google scholar] [PubMed]
- Yao P, Lin P, Gokoolparsadh A, Assareh A, Thang MWC, Voineagu I. Coexpression networks identify brain region-specific enhancer RNAs in the human brain. Nat Neurosci. 2015;18:1168-1174. [CrossRef] [Google scholar] [PubMed]
- Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46:881-885. [CrossRef] [Google scholar] [PubMed]
- Schaaf CP, Sabo A, Sakai Y, Crosby J, Muzny D, Hawes A, et al. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum Mol Genet. 2011;20:3366-3375. [CrossRef] [Google scholar] [PubMed]
- Wall DP, Kosmicki J, Deluca TF, Harstad E, Fusaro VA. Use of machine learning to shorten observation-based screening and diagnosis of autism. Transl Psychiat. 2012;2:e100. [CrossRef] [Google scholar] [PubMed]
- Krishnan A, Zhang R, Yao V, Theesfeld CL, Wong AK, Tadych A, et al. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat Neurosc. 2016. [Google scholar]
- Krumm N, O’Roak BJ, Karakoc E, Mohajeri K, Nelson B, Vives L, et al. Transmission disequilibrium of small CNVs in simplex autism. Am J Hum Genet. 2013;93:595-606. [CrossRef] [Google scholar] [PubMed]
- McCarthy MM, Wright CL. Convergence of sex differences and the neuroimmune system in autism spectrum disorder. Biol Psychiat. 2017;81:402-410. [CrossRef] [Google scholar] [PubMed]
- Schaafsma SM, Gagnidze K, Reyes A, Norstedt N, Månsson K, Francis K, et al. Sex-specific gene–environment interactions underlying ASD-like behaviors. P Natl Acad Sci. 2017:201619312. [Google scholar]
- Werling DM, Parikshak NN, Geschwind DH. Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nature Commun. 2016;7. [CrossRef] [Google scholar] [PubMed]
- Diaz-Beltran L, Esteban FJ, Varma M, Ortuzk A, David M, Wall DP. Cross-disorder comparative analysis of comorbid conditions reveals novel autism candidate genes. BMC Genomics. 2017;18:315. [CrossRef] [Google scholar] [PubMed]
- Kirov G. CNVs in neuropsychiatric disorders. Human molecular genetics. 2015;24(R1):R45-R49. [CrossRef] [Google scholar] [PubMed]
- Pescosolido MF, Gamsiz ED, Nagpal S, Morrow EM. Distribution of disease-associated copy number variants across distinct disorders of cognitive development. J Am Acad Child Psy. 2013;52:414-430. [CrossRef] [Google scholar] [PubMed]
- Kou Y, Betancur C, Xu H, Buxbaum JD, Ma'Ayan A, editors. Network‐and attribute‐based classifiers can prioritize genes and pathways for autism spectrum disorders and intellectual disability2012: Wiley Online Library. [Google scholar]
- McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiat. 2014;19:652-658. [CrossRef] [Google scholar] [PubMed]
- Kim DS, Burt AA, Ranchalis JE, Wilmot B, Smith JD, Patterson KE, et al. Sequencing of sporadic Attention‐Deficit Hyperactivity Disorder (ADHD) identifies novel and potentially pathogenic de novo variants and excludes overlap with genes associated with autism spectrum disorder. Am J Med Genet B. 2017. [Google scholar]
- Lo M-T, Hinds DA, Tung JY, Franz C, Fan C-C, Wang Y, et al. Genome-wide analyses for personality traits identify six genomic loci and show correlations with psychiatric disorders. Nat Genet. 2016. [Google scholar]
- Talkowski ME, Ernst C, Heilbut A, Chiang C, Hanscom C, Lindgren A, et al. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am J Hum Genet. 2011;88:469-481. [CrossRef] [Google scholar] [PubMed]
- Ruaud L, Mignot C, Guët A, Ohl C, Nava C, Héron D, et al. DYRK1A mutations in two unrelated patients. Eur J Med Genet. 2015;58:168-174. [CrossRef] [Google scholar] [PubMed]
- MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469-476. [CrossRef] [Google scholar] [PubMed]

