

Temporal Lobe Epilepsy, Stroke, and Traumatic Brain Injury: Mechanisms of Hyperpolarized, Depolarized, and Flow-Through Ion Channels Utilized as Tri-Coordinate Biomarkers of Electrophysiologic Dysfunction
Gina Sizemore 1,*![]() , Brandon Lucke-Wold 2
, Brandon Lucke-Wold 2![]() , Charles Rosen 2
, Charles Rosen 2![]() , James W. Simpkins 3
, James W. Simpkins 3![]() , Sanjay Bhatia 2
, Sanjay Bhatia 2![]() , Dandan Sun 4
, Dandan Sun 4![]()
-
Department of Clinical and Translational Science, West Virginia School of Medicine, Morgantown, WV
-
Department of Neurosurgery, West Virginia School of Medicine, Morgantown, WV
-
Center for Basic and Translational Stroke Research, West Virginia School of Medicine, Morgantown, WV
-
Department of Neurology, University of Pittsburgh, Pittsburgh, PA
* Correspondence: Gina Sizemore ![]()
Academic Editor: Bart Ellenbroek
Received: January 12, 2018 | Accepted: May 20, 2018 | Published: June 4, 2018
OBM Neurobiology 2018, Volume 2, Issue 2, doi:10.21926/obm.neurobiol.1802009
Recommended citation: Sizemore G, Lucke-Wold B, Rosen C, Simpkins JW, Bhatia S, Sun D. Temporal Lobe Epilepsy, Stroke, and Traumatic Brain Injury: Mechanisms of Hyperpolarized, Depolarized, and Flow-Through Ion Channels Utilized as Tri-Coordinate Biomarkers of Electrophysiologic Dysfunction. OBM Neurobiology 2018;2(2):009; doi:10.21926/obm.neurobiol.1802009.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
The brain is an integrated network of multiple variables that when compromised create a diseased state. The neuropathology of temporal lobe epilepsy (TLE), stroke, and traumatic brain injury (TBI) demonstrate both similarity and complexity that reflects this integrated variability; TLE with its live human tissue resection provides opportunity for translational science to demonstrate scale equivalent experimentation between the macroscopic world of clinical disease and the microscopic world of basic science. The extended value of this research is that the neuroinflammatory abnormalities that occur throughout astrocytes with hippocampal sclerosis and damaged or even reversed signaling pathways (inhibition to excitation such as with gaba-aminobutyric acid) are similar to those seen in post-stroke and TBI models. In evaluation of the epilepsy population this interconnectedness of pathology warrants further evaluation with collaborative efforts. This review summarizes patterns that could shift experimentation closer to the macro level of humanity, but still represent the micro world of genetics, epigenetics, and neuro-injury across etiologies of physiologic dysfunction such as TLE, stroke, and TBI with evaluation of cell function using electrophysiology. In conclusion we demonstrate the plausibility of electrophysiologic voltage and current measurement of brain tissue by patch clamp analysis to specify actual electrophysiologic function for comparison to electroencephalography in order to aid neurologic evaluation. Finally, we discuss the opportunity with multiscale modeling to display integration of the hyperpolarization cyclic-nucleotide gated channel, the depolarized calcium channels, and sodium-potassium-chloride-one/potassium-chloride-two co-transporter channels as potential mechanisms utilized as tri-coordinate biomarkers with these three forms of neurologic disease at a molecular scale of electrophysiologic pathology.
Keywords
Epilepsy; ion channels; electrophysiology; neurologic diseases
1. Introduction
Recent statistics indicate 50 million people suffer with epilepsy [1]. In adults with focal epilepsy, about 50% of them have had a previous brain injury [2]. Approximately 2.3% to 14% of stroke patients will develop seizures [3] and temporal lobe epilepsy (TLE) develops at rates inconsistent with heritability alone lending credence to underlying brain damage or otherwise dysfunction as a contributing feature [4]. A common feature in TLE, stroke, and traumatic brain injury (TBI) is the electrophysiologic dysfunction. This review will utilize the common thread of electrical biophysics to identify existing knowledge, expose the gaps, and project a plan for future research.
Although the pathophysiology of epilepsy often begins with the discussion of genetic mutations, such as the SCN1A gene mutation with over 350 identified mutations [5], those mutations are not consistent across all epilepsy. For example, a specific mutation in the SCN1A gene, which affects an alpha1 subunit in the voltage-gated sodium channel, is not compromised in all epilepsies and not researched at all in diseases like stroke or TBI. Yet, SCN1A has demonstrated a phenotype associated with hemiplegic migraine [6], which is often a symptom in TBI. Newer research, at smaller scales, reveals mutations of a variety of genes that encode for signal transduction, synaptic transmission, neuronal metabolism and excitability, as well as brain development that play a role in disease pathology. In addition, variability in epigenetics distributes change that do not alter the original DNA, but create a disease state by alterations like methylation, and histone modification that can silence or overexpress proteins. Genes can be evaluated with specificity of their protein expression at the tissue injury region. These epigenetic contributors are demonstrated across a wide range of epilepsies and show correlation with encephalopathy seen across the neurophysiological and biophysical mechanisms of blood brain barrier compromise in stroke, ion channelopathy of TLE, and white matter apoptosis and necrosis of TBI. This review builds a plan to pull up from the microscopic and pull down from the macroscopic to define a place in the middle using a scale of disease evaluation that can be communicated between the discovery of basic science and the application of clinical medicine.
Despite this wealth of knowledge, greater than 30% of epilepsy patients continue to battle with a drug resistant seizure disorder while stroke and TBI patients bear the persistent symptoms related to the electrophysiologic dysfunction that ultimately reflects neurodegenerative change of varying degrees. Neuropathology of electrophysiologic dysfunction is complex and encompasses neuro-injury, epigenetics, and genetics. This complexity requires organization of scale to evaluate the interdependence and integration of these components. The excitability and inhibition balance required for homeostasis can be compromised by signaling cascades, genetics, or system failure across multiple neuronal circuits [7]. Therefore, selecting three diverse forms of electrophysiologic dysfunction: stroke, traumatic brain injury, and temporal lobe epilepsy - this review will discuss their commonality of electrophysiologic dysfunction, the limitations of the tools for evaluating electrophysiologic dysfunction, and the benefit of focused analysis with a hyperpolarized (HCN) channel, a depolarized calcium (CaV) channel and the sodium-potassium-chloride-one/potassium-chloride-two (NKCC1/KCC2) co-transporters. They can be correlated with the epigenetic expression, RNA and protein analysis to determine biomarkers for improved evaluation and targeted drug treatment of these diseases.
2. Translational Research
A challenge in translational research is application of precise, controlled basic science research to the multi-variable clinical reality. Therefore, evaluation across the three diseases of stroke, TLE, and TBI demonstrates how electrophysiology offers a broad level translational step between the complexity of the disease (clinical) and the specificity of individual dysfunctional components (basic science). This review utilizes location, morphology, and neuropathology to create a context for the discussion.
Location: Circulation to the brain arises from both the internal carotid arteries anteriorly and posteriorly through the vertebrobasilar artery system [8] . The hippocampus, the focal region of TLE, is fed through the posterior system with anterior contribution from the anterior choroidal artery. Although, not the only contributor to translational research shortcomings from mouse to clinic the vascular diversity requires consideration. The mouse does not evidence the anterior choroidal artery seen in humans, but with a smaller surface area reflects a more collateral system compared to the more end arterial system of humans. In Figure 1 you see the comparison of the mouse and human vascular system. The mouse brain (1A) is created by a novel perfusion technique that allows resin casting of the mouse vasculature that maintains exact measurements of vascular diameter and length while eroding all other brain tissue to leave only a model of the vascular system, no brain tissue. In addition, the casting system maintains the network connections to demonstrate the collateral system of vasculature in the mouse brain. Everything in the picuture is vasculature. Compare that to the perfusion of the human brain (1B) in CT scan analysis. The end arterial system is a striking contrast to the more collateral system seen in the mouse vasculature. In (1C) the anterior choroidal artery is marked, as it is the only artery reaching to the hippocampus from the arterial system in humans, apart from the recurrent artery of heubner. Again, this is contrasted to the more collateral mouse vasculature to the hippocampus, which may afford it some neuroprotection during injury. It is important to continue animal model research. However, it is paramount that research of human tissue at this scale be developed to offset the limitations of animal models and build on their successes.
Major injury in stroke most frequently occurs from the anterior system, while TBI injury more frequently involves the posterior system through vertebrobasilar dissection. Therefore, TLE offers the opportunity of a control disease by virtue of consistent hippocampal location injury for evaluation with the variant anterior-posterior hippocampal injury from stroke and TBI. The anterior choroidal artery, a branch off the internal carotid, just distal to the posterior communicating artery is part of the anterior circulation, which runs deep to the hippocampal region. In addition, the recurrent artery of heubner originates from the anterior system. The variation between these two structures is consistent in that when one is dominate the other balances to a smaller development. The anterior choroidal represents part of the residual circulation from our evolutionary past before the posterior circulation developed with our higher cortical brain and therefore along with the heubner artery offers an opportunity to compare and contrast data with a mouse model in the evaluation of stroke, TBI, and TLE. Though absence of the anterior choroidal and heubner artery in mice is not the singular cause for the translational difficulty, research can demonstrate key functional variances between a collateral system (mouse) and an end artery system (human) in the circulation within a unique vascular region of brain.
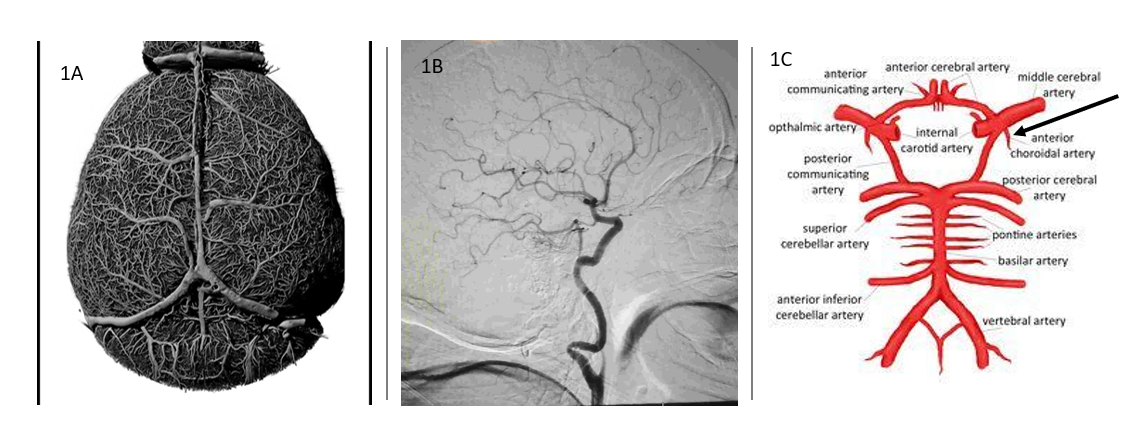
Figure 1 The casting of the entire vascular network 1A in a 24-month old male wild type mouse. The cast created with polyurethane resin and image acquired with micro CT system for visualization of collateral vasculature structure (Picture courtesy of Dominic Quintana, Neuroscience Department, West Virginia University). In 1B Computed Tomography Angiography of the normal single side internal carotid end artery system in human. The internal carotid artery supplies the anterior circulation to the brain and includes the middle cerebral artery (MCA) commonly associated with stroke and the anterior choroidal artery, which is a feeding vessel, from the anterior system, the hippocampal region associated with the sclerosis of temporal lobe epilepsy. In 1C the anterior choroidal artery in humans is the only artery branch feeding the hippocampus from the anterior system, apart from the recurrent artery of heubner. The mouse has a more collateral reach to the hippocampus possibly contributing some neuroprotection during injury.
Approximately two-thirds of ischemic stroke arises in the anterior circulation with 90% of those involving infarct of the MCA [9]. However, despite multiple animal models of MCA injury, we have failed to discover new drug therapy or targets that translate to successful clinical application. The anterior choroidal artery, which lies proximal to the MCA and the end artery structure of humans contrasted to the more collateral structure of mice may offer clues as science develops improved biotechnology to traverse these diverse pathways. In TBI, the most common vascular injury is subarachnoid haemorrhage [10] with secondary vascular issues from vasospasm or lumen narrowing of arteries from edema or increased intracranial pressure that further reduce brain perfusion [11]. Vertebrobasilar dissection is more common than anterior dissection in traumatic brain injury. The posterior vasculature diameter is narrow and causes difficulty, even under more stable situations such as bypass grafts where new techniques have been developed to overcome the disadvantage of these high-flow grafts due to the mismatch in vessel diameter between donor and recipient vessels in the posterior circulation [12]. With the hippocampal sensitivity to ischemia dissection in the posterior system leads to decreased perfusion by both blood loss and arterial narrowing due to acute spasm. The hippocampus is sensitive to ischemia and periods of excessive intracranial pressure and blood loss could demonstrate more significant injury in an end arterial human system compared to a more collateral system. Finally, TLE, which is noted for its hippocampal sclerosis, remains devoid of significant vascular evaluation despite our knowledge of glial cell function in maintenance of the blood brain barrier and its dysfunction in developing sclerosis. The brain vasculature represents the pathway in and out of the neurologic circuitry and is too broad to cover the entire network for all three diseases. Therefore, location will be restricted to the vascular distribution of the hippocampus which reaches deep into our evolutionary past, yet still offers parameters with defined limits for the discussion of stroke, TBI, and TLE within a framework that makes them consistent with one another by virtue of location. In addition, it addresses specific gaps in our knowledge of glial involved vascular injury in TLE, hippocampal role in cognitive and memory decline in TBI, and isolates a specific region for analysis of core versus penumbra injury in stroke. “The penumbra is characterized by the loss of action potential firing but maintenance of proper resting membrane potential” [13]. Resting membrane potential and action potential firing are key electrophysiologic targets translational research can utilize.
Electrophysiology: Neuroimaging is the gold standard evaluation in stroke and TBI and offers benefit in surgical planning with TLE. However, despite the benefits of these tools, they fail to establish data that connects structural abnormality and electrophysiologic dysfunction in the brain that translates to improved clinical outcomes [14]. EEG provides cortical activity markers in real-time [15] which augment the time lag in neuroimaging. However, few facilities have compatible tools to perform these tests synchronisly and have led to decreased use of electrophysiologic tools in preference to BOLD MRI, perfusion CT, and specialized manipulations of scans as accessories to primary MRI and CT for many neurologic diseases. Therefore, assessment of neuronal oscillations with electroencephalography and ion channel function is a novel way to assess the balance between excitatory and inhibitory cortical processes and provide biomarkers for future evaluation and treatment.
The HCN1 channel, the CaV channel, and the NKCC1/KCC2 cotransporter channels are representatives of hyperpolarization, depolarization, and flow through that can create a three-prong coordinate evaluation of current that can be used as a biomarker to aid evaluation and treatment being done with neuroimaging and electrophysiology tools such as EEG. In addition, it reflects the opportunity for bioengineering to develop alongside new electrophysiologic technologies such as microelectrode arrays, which improve spatial resolution over EEG. These data can be correlated with data from electrophysiology (See Figure 2). The glass pipette patching an astrocyte (2C) indicates the opportunity electrophysiology offers to bring the macroscopic world (2A) the glass pipette electrode and (2B) the microscopic world of an astrocyte together and produce (2D) research data in real time electric current evidence of cell function that communicates with a language that is the same in the macroscopic and microscopic world. Thus, creating the possibility to translate information between scales of the very small and precise (basic science discovery) and scales of the very large and complex (clinical application).
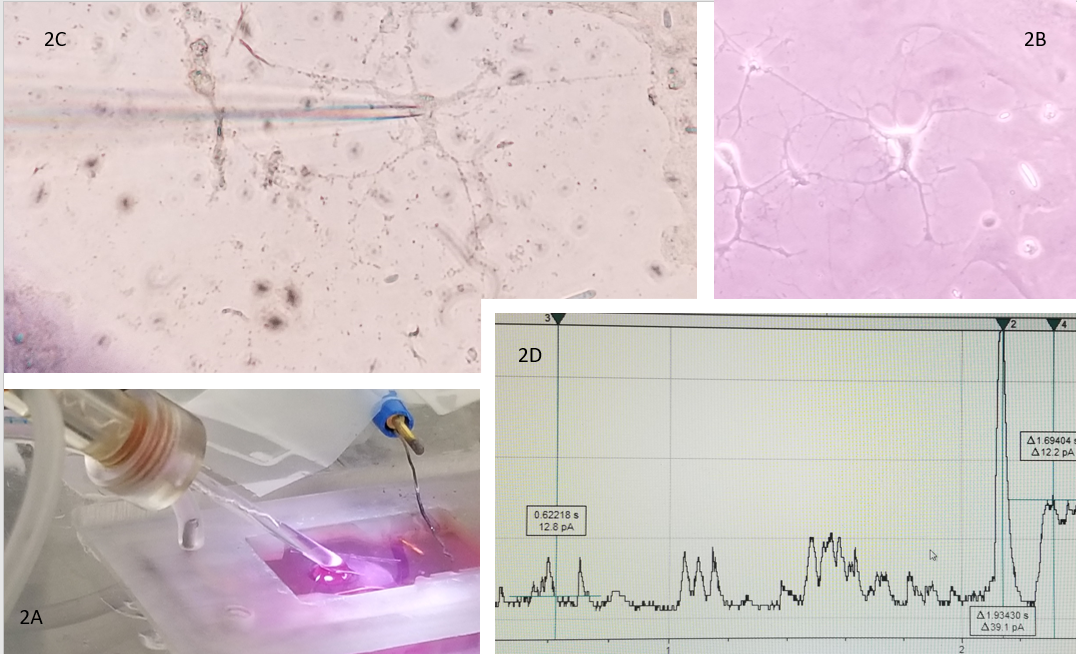
Figure 2 The glass pipette as electrode 2A in the recording bath where the astrocyte 2B is adhered to the coverslip. 2C demonstrates the patch between the electrode and the astrocyte under the microscope. Finally, 2D is an example of real time electrical current received from a cell.
Morphology: Morphology evaluation is based on literature, which demonstrated large-scale involvement of astrocytes and pyramidal neurons in all three diseases. Astrocytes are an active and reactive component of the brain that influences gene expression, extracellular matrix components, and metabolism. When injured they display distinct features compared to healthy astroglia in their morphology, expansion, and signaling in ischemia [16] as well as TBI [17], and sclerosis in TLE [18]. Pyramidal neurons are the most common recipient within the environment of change commanded by astrocytes. Specific cell types vary by location which ultimately effects their function within those regions and the hippocampus remains a controversial region [19]. In addition, ion channel distribution on any given cell differs and contributes variation to the cell function. The pyramidal neurons of the CA1 region of the hippocampus are extremely sensitive to ischemia and ion channel research continues to evaluate this for mechanism though it is not yet well defined [20]. Unlike the L5 pyramidal neurons, which stretch through all five levels of the cortex, the CA1 pyramidal neuron structures remain confined to the CA1 hippocampal region and offer a specific region for discussion and experiment to isolate changes affected by stroke, TBI, and TLE. The diversity of cell structure contributes to the variety of function each cell offers to the brain (See Figure 3). It demonstrates the variance of a pyramidal neuron (3A); notice the CA1 and CA3 pyramidal neurons, of specific interest within the hippocampus, compared to the Layer V(LV) pyramidal neuron. In (3B) the LV pyramidal neuron with its streamline look reaches through all six layers of the neocortex, with little to no branching, while the more compact CA1 and CA3 of the hippocampus with their apical branching are designed for their local purpose. Research indicates a large percentage of HCN channels are expressed in the apical region of pyramidal neurons. The benefit of whole cell patch is it reads the resting membrane potential of the entire cell at the soma (3C). The alternate protocols available in whole cell patch, such as Ramp protocol (3D) and Pulse protocol (3E) allow evaluation of specific electrical changes in the entire cell. Therefore, the hippocampus, with the apical girth of CA1 and CA3 offers opportunity for significant evaluation with electrophysiology of these hyperpolarized channels in epilepsy and other neurologic disease.
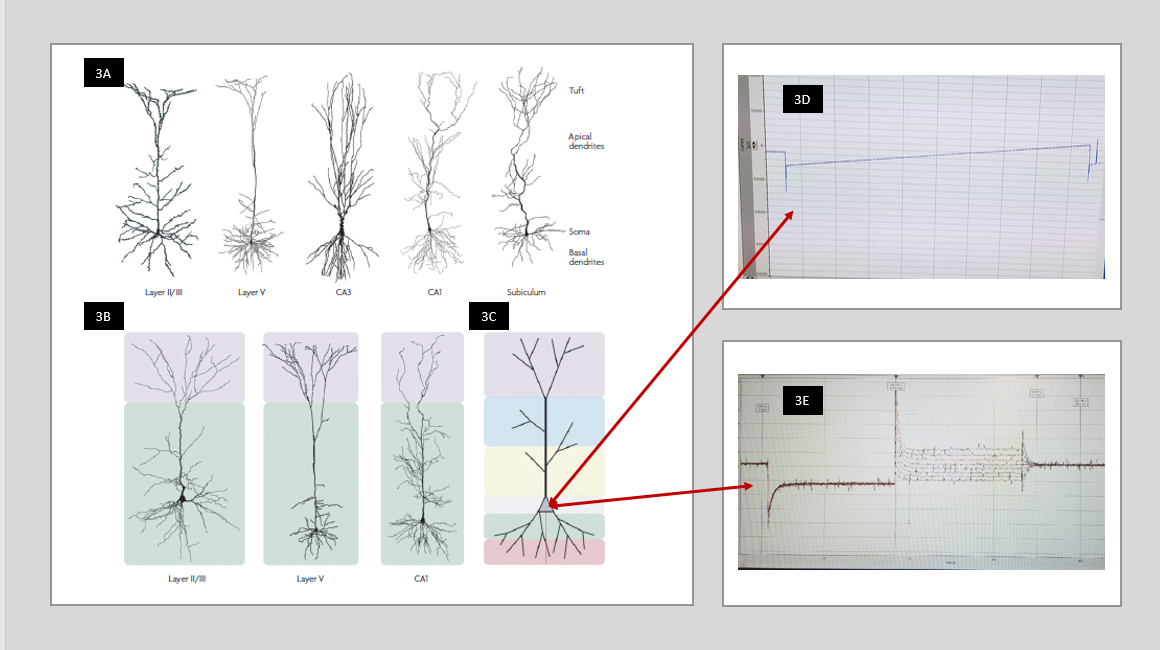
Figure 3 In 3A the variability of the pyramidal cells is shown by the CA1 and CA3 pyramidal neurons compared to the Layer V (LV) pyramidal neuron when you look at where and how the branching occurs. The LV pyramidal neuron has a streamline look and reaches through all six layers of the neocortex 3B while the more compact CA1 and CA3 of the hippocampus with their apical branching are designed for their local purpose. In 3C the arrows indicate the location of the soma where patch clamp recording gives the reading of the resting membrane potential of the entire cell. Picture reprinted by permission from Macmillan Publishers Ltd: [Nature Reviews Neuroscience] [21], copyright (2008). Finally, in 3D, the ramp protocol, and 3E, the pulse protocol, you see examples of recordings received from a cell with two different protocols in the whole cell patch technique. Protocol examples are from our research lab at West Virginia University.
Genetic and epigenetic modifications play a role in the channel construction, and expression of the channels discussed in this review. Within the spectrum of each disease the full context of those modifications is not yet understood. This review’s purpose is to demonstrate the pathology based on the role of the channels within the cell for depolarization, hyperpolarization, and flow through or electroneutral current to open further discussion of experiment direction for disease specific research evaluating these channels as coupling or co-contributing channels in multiple disease pathology. HCN as a hyperpolarizing channel leads to hyperexcitability when it is mutated from its normal function, Ca leads to multiple variations of depolarization and intracellular activity when it mutates from its normal function, and the electroneutral cotransport channels already demonstrated as coupling with sodium and potassium alter their flow-through neutral status when they mutate from normal function. Approaching this from a predominantly electrophysiologic perspective puts focus on the function of the channels when they are mutated or have variant expression more than on the depth of discussion of each mutant variation. A mutation either causes the channel to work more effectively, less effectively, or not at all. Overexpression or underexpression similarly may impact function, but may also not affect cell function at all. This review aims to discuss the role of these channels in the broader terms of cell function in the context of disease rather than specific mutations for any gene family.
Figure 3D Demonstrates a Ramp protocol, which is the simplest screening tool to evaluate channels that open with depolarization. We have used this protocol in research to demonstrate how overexpression does not always tell you what the function will be in the cell. If there is overexpression of a specific channel on a scale of 1000 to 1, yet only 5% of channels are open at the physiologic state then having overexpression of essentially 1000 doors does not matter. The function in the cell does not change if they are not open. Therefore, electrophysiology can give us functional data about what is going on in the cell without being distracted by genetic mutations that may pose no effect to the function or dysfunction of the cell in the disease. Yet equally, when there is abnormal function in the cell researchers can track that dysfunction back to its specific components of mutation and expression.
Neuroinflammation: The signaling pathways for neuroinflammatory response have been exposed as a contributor to pathology in neuro trauma, whether the acute injury of stroke, TBI, or the chronic sclerosis of TLE. Research linked NKCC1 signal increase to increased ischemic risk [22]. Inflammation of astrocytes, seen in temporal lobe epilepsy, occurs due to excess glutamate through the glutamine/glutamate/cycle (GGC) [18]. There is significant change demonstrated in the pathway from epileptogenesis through sclerosis in a hippocampal astrocyte cell. Comparable astrocyte damage occurs in post-stroke by compromise of the arteries where astrocytes assist in maintaining the blood-brain barrier and in TBI with similar compromise of the blood-brain barrier where in mouse models it is associated with memory changes [23]. There is glutamate uptake at the astrocyte where it is converted by glutamine synthetase to glutamine and released to the extracellular space. The neuron then uptakes the glutamine where it is degraded to glutamate by phosphate activated glutaminase (PAG) and released back to the extracellular space as glutamate to continue the cycle. There are multiple opportunities for the system to fail in this cycle from glutamine synthetase deficiency or glial glutamate (GLT-1) transporter dysfunction in the astrocyte to neuron dysfunction by excessive release due to (PAG) and the excitatory amino acid carrier (EAAC1); all of which contribute to extracellular/intracellular ion gradient shifts that disrupt homeostasis.
Neurodegeneration: Neurodegeneration is a natural path from the epigenetic contributions of neuroinflammation. The degeneration of neural tissue in neurologic disease is not singular in its origin and due to mixed contributions of neuroinflammation, gene expression, and ion dysregulation it is difficult to determine the cause and direction of pathology [24]. The epigenetic impact of the inflammatory process can create signal error or excess that leads to sclerosis and/or blood brain barrier breach [25]; just to name a few. New research indicates mitochondrial mechanism with miR-34 mediated regulation of the blood brain barrier and analysis with bioinformatics indicates several gene targets [26]. Research correlating upregulation and down regulation of co-transporter channels such as NKCC1 and KCC2 respectively following injury [27] support research of these channels in the pathology of stroke, TLE, and TBI where neuro inflammatory processes led to neurodegeneration. In addition, inflammation can produce neurodegeneration by abnormal gene expression, trigger regulating error, and channel surface expression variation with post-translational modification. For example, the HCN1 channel has an exposed extracellular post-translational modification where circulating inflammatory signals may contribute to dysfunction. Further review of a susceptible post-translational modification site exposed on the HCN1 channel can be found in Lee et al., 2017 [28]. These epigenetic influences of neuroinflammation can diverge from neurodegeneration and impact neurogenesis as well.
Neurogenesis: Increased ion channel density demonstrated significantly depolarized astrocyte cells in hippocampal foci damaged regions (~55 mV) compared with astrocytes in hippocampal neurotypical regions (~75 mV) since early channel research [29]. This channel density neurogenesis continues in recent research, most notably in new research regarding NKCC1 and KCC2 co-transporters where ratio shifts to embryologic conditions indicate again the role of channel density in the probability for dysfunction [30]. In addition, the electrophysiologic effect of channel density on surface expression, post-translational modification, and signaling demonstrate the interconnectedness and circular difficulty of the pathology. The anticipated voltages can be calculated by Nernst equation demonstrating the polarity shift that the ratio inversion of NKCC1 to KCC2 could create in the membrane potential (see Figure 4). The Nernst equation in figure four demonstrates the role that concentration of a single ion can play in altering the membrane potential of a cell. With so many basic science variables from gene expression to neuroinflammation, neurodegeneration, and neurogenesis it is necessary to find a system that reflects the function of them all. Electrophysiology can extract the functional data that in turn can be correlated with microscopic science and macroscopic disease.
3. Ion Channel Milieu
Regulation of ion homeostasis is critical to the function of cells. Ion transporters establish that by cell volume, voltage influence of membrane potential, and signal transduction [31]. Shifts in cellular volume and gradient pressures trigger homeostatic responses coordinated by ion channels to mediate and avoid excessive cellular edema or dehydration with regulatory volume adjustments [32]. Membrane potential is primary in the discussion of cell survival and function. Therefore, membrane potential is the necessary starting point. The HCN1 channel with its Ih funny current displays a reversed polarity in its biophysical nature with a reversal potential ~ -20 mV carrying a cation current of both Na and K that is inward moving at rest [33]. This combination along with additional features to be discussed creates an intersection of opportunity for interaction with sodium-potassium-chloride co-transporters NKCC1/KCC2. Signal transduction pathology disrupts cell homeostasis. It is here where ion channels can create significant influence. GABA is one of the most researched contributors to signaling pathways and demonstrates the importance of ion channel interaction in the discussion of negatively charged ion concentrations, such as Cl- within the cell membrane [34].

Figure 4 Nernst equation calculation for Chloride demonstrates the electrophysiologic alteration of membrane potential possible with fluctuations of Cl- in conditions such as ratio shift of NKCC1 to KCC2. Their correct ratio is necessary for neutrality of charge. Repeat the calculation on your own by changing the outside and inside molarity to demonstrate what can happen if the ratios of expression for these channels are not appropriate.
4. Hyperpolarized Cyclic-Nucleotide Ion Channels
4.1 Sequencing, Construction, and Function
HCN role in epilepsy is a function of its isoform’s homomeric or heteromeric channel construction, expression, and interaction. There are four HCN isoforms: HCN1, HCN2, HCN3, HCN4. The expression of HCN1 is more prevalent in the hippocampus and cortex compared with the HCN2, and HCN4 isoforms, which are more prominent in the thalamus and sinoatrial node [35]. In discussing the role of HCN channels in epilepsy it is not a singular mutation of the channel but rather a combination of channel construction variance as well as altered expression which demonstrated epileptic disease in mouse models [35]. In addition, deletion of HCN1 and HCN2 in knock-out mice created seizure prone mice [36].
However, the structure of the HCN1 channel is showing significant promise for integrated roles with its reversed polarity [37]. The hyperpolarization as opposed to depolarization gating make the HCN1 channel an excellent candidate for integrated research with NKCC1 and KCC2 co-transporter channels along with the depolarization gating of the high voltage-gated CaV channels. The construction of the HCN1 channel pore has great conservation with that of the K channels; both contain the glycine-tyrosine-glycine (GYG) as a selectivity filter [38]. The function of the HCN1 channel is to operate with reverse polarity and it does this by structural design that allows for conformational changes of the pore based on the trigger mechanism. Using cryo-electron microscopy (Cryo-EM), researchers demonstrated the alternated gating between hyperpolarization and cAMP modulation [37]. We hypothesize that hyperpolarization channels like HCN1 in conjunction with inappropriate NKCC1/KCC2 co-transport channel ratios allows function gain or inhibition at a more negative membrane potential leading to hyperexcitation, epileptogenesis, neuroinflammation and therefore has a high potential link to hippocampal astrocytic and pyramidal neuron injury with evidence of electrophysiologic dysfunction either as neurodegeneration, hyperexcitation or both in stroke, TBI, and TLE. HCN produces a current called funny current, If, in the heart and is considered the pacemaker current. It is often written as Ih when discussing its current in the brain. The Ih current is an inward current [39]. Its complexity of construction from homomeric to heteromeric, as well as expression of the channel, impacts how closely to resting cell membrane potential the channel can be triggered. Additionally, reduction in expression as well as complete deletion such as is done in knockout mice consistently indicates increased seizure risks and activity in other neurologic disease such as stroke and TBI.
Channel electrophysiology connects with epilepsy, stroke, and traumatic brain injury by offering a context of functional evaluation to these diseases. Each disease has pathology rooted in failing cellular processes of which ion channels are a key component. When using an evaluation of scale as macroscopic as a complete human disease it is difficult to correlate it with equal scale at the basic science level (i.e. discuss the function of a cell, as a whole, rather than a single variable of change such as a histone modification that changes the expression of a gene). Each of these individual discoveries is important. However, electrophysiology of cell function creates a closer equality of scale for translational science because the microscopic data is measured, discussed, and analyzed with the same tools as the macroscopic data. Resting membrane potential, depolarization, and hyperpolarization states are mandatory tools for a cell’s homeostatic balance. In this review we press for the whole homeostatic electrical balance system of the cell to be used for representation of the larger scale human disease. It is not equivalent, but it is a starting point to begin the translational discourse between clinical and basic science. Ion channels play a role in the diverse signaling throughout stroke injury at glial cells, neurons, and the blood brain barrier [40]. Traumatic brain injury leads to variant excitotoxicity, which could be evaluated, with electrophysiology to better understand the functional changes at the cellular level [41]. Epilepsy results in hyperexcitability recorded by electroencephalography and confirmed in electrophysiology recording baths at the cellular level by evaluation of ion channel function [42]. Experimentation to evaluate these diseases for similarities with channels specific for these electrical potentials can establish a baseline for deeper discovery of the disease process. Electrophysiology is the translational lane of research that is yet to be completely developed and explored; a lane that runs like a blood brain barrier between the worlds of clinical research and basic science (See Table 1). This electrophysiologic diversity can be evaluated further with future research since evidence for all four HCN isoforms in the brain have been described [43].
Table 1 The voltage measurements from Della Santina [44] in their work with HCN1 channels in the retina converted to a table to demonstrate the value of monitoring Ih current of the HCN1 channel. The results indicate the channel in response to hyperpolarization and depolarization and indicate the potential for biomarker mechanism in the electrophysiologic function of neurologic disease.

5. NKCC1 and KCC2 Co-Transporter Ion Channels
5.1 Sequence, Construction, and Function
Research on variants have associated R952H and R1049C of the KCC2 co-transporter channel with idiopathic generalized epilepsy (IGE) [45] a more generalized epilepsy. KCC2 plays a dominant role in normal development, influencing factors such as the excitatory-inhibitory signal read of GABA [46]. When KCC2 is deficient, especially in relevance to NKCC1, brain development is compromised. Intracellular chloride currents show biophysical accumulation when the ratio construction of NKCC1 expression is higher compared to KCC2 expression [47]. This leads to severe neurologic dysfunction and delay noted due to decreased function of the NKCC1/KCC2 co-transporter’s ability to maintain low intracellular Cl- because of mutations at SLC12A5 [48]. The fundamental role of Na-K-Cl cotransporter 1 (NKCC1) and K-Cl cotransporter 2 (KCC2) is lost when the expression of each is not regulated properly. In addition, the integrated signaling system as it relates to GABA is disrupted [49]. The NKCC1 and KCC2 provide the Cl- homeostasis voltage within the cell [50], which is why they are pertinent to evaluation of hyperpolarization-gated channels like HCN1.
The deregulation of NKCC1 in epilepsy is demonstrated by the increased association with epileptogenesis due to its role in managing the concentration of intracellular chloride [51]. The slc12 gene family encodes both NKCC1 and KCC2 and mutations in this gene have been associated with epilepsy. Many of these electroneutral chloride cotransporters functionally couple movement of chloride with Sodium and potassium and have demonstrated pathological hyperexcitability in mice with genetic modifications of this gene [52].
In models of evaluation these chloride channels demonstrate the detrimental effect of neuroinjury on their ratios to one another. Multiple research platforms including ischemic stroke, TLE, and subarachnoid hemorrhage (the most common brain injury in TBI) reveal the correlated role between disease and damage in these channels. Western blot analysis by Yong Tien, et al (2015) revealed multiple neuroinflammatory components following subarachnoid hemorrhage. The NKCC1 and KCC2 ratio shifts were significant, demonstrating changes of their protein expression [53]. Western blot analysis characterized a decline in KCC2 expression and increase expression in NKCC1 following induced subarachnoid hemorrhage in their rat model. Topiramate, a frequently used anticonvulsant, effectively reversed those KCC2/NKCC1 expressions in a dose-dependent fashion. This response is a positive indicator of pharmacologic opportunity as we learn more about the integration of these channel functions.
6. High Voltage-Gated Calcium Ion Channels
High voltage-gated calcium ion channels are discussed briefly below due to their connections to epilepsy and the role they could play in the development of an integrated computational model of ion function for future research.
HVA calcium channels have five known types, all derived with an original subunit: CaV1 the L-type, CaV2 including the P/Q-type [CaV2.1] the R-type [CaV2.2] and N-type [CaV2.3], and the CaV3 the T-type. These types have been studied with regards to genome structure, mutation, pathology, and pharmacology. The results thus far are promising. The P/Q type is the CaV2.1 type of the five types and has the most connection with epilepsy along with the CaV3.2 T-type. They both are linked to IGE. The HVA channel requires higher depolarization, compared to LVA channels. Yet, both are linked to IGE, both are pharmacologically responsive, and have improved epilepsy outcomes with inhibition in patients [54]. Low Voltage Gated Channels like the T-type channels offer the first glimpse of opportunity for hyperpolarized channels and negative charge transporters to be linked to IGE. The structural changes and pathology of these channels is tied to congenital mutations associated with IGE which raises the question for susceptibility to more epigenetic, neuroinflammatory changes linked with more focal epilepsy like TLE, post-stroke epilepsy (PSE), and post-traumatic epilepsy (PTE). The voltage-gated channels have many polymorphisms that contribute to the mutations leading to structural and functional change [55], but alone they do not answer all the questions regarding electrophysiologic dysfunction.
The CaV families, which are the L-type channels [CaV1], P/Q-type [CaV2.1] (R-type and N-type of CaV2 family not reviewed here), and T-type [CaV3] channels, have defined structure. The CaV1 and CaV2 types have an alpha subunit that forms the pore of the channel and a gamma, beta, and alpha2delta unit forming the tertiary structure. These channels are closely related to the CaV3 channel, which also has an alpha unit pore [56]. Splicing variants of each subunit contribute to altered phenotypes associated with epilepsy [57]. These splice variants determine biophysical properties and tissue location of channels [58].
Current data from both animal models and clinical trials point to CaV2 the P/Q type HVA channel, and the CaV3 T-type LVA channel as important, but not alone in their epileptogenesis role. These channels are well studied and presented briefly here due to their strong link to IGE that could shed light on the genetic contributions to the electropyysiologic dysfunction of TLE, stroke, and TBI. In addition, the role of calcium in contributing to glutamate excitotoxicity is relevant [38]. Whether from ischemia, TBI, or epilepsy the rich research database on HVA calcium channels can help form a background for baseline computational modeling regarding ion gradients.
7. Clinical Relevance and Conclusion
The animal models for stroke, TBI, and TLE are beneficial to science and humanity in our growth of knowledge, yet they often fail to translate to improved outcomes in human clinical applications. Animal models have demonstrated multiple failures in producing drugs with safety and efficacy in humans despite success in preclinical experiments [59]. What are the limitations that contribute to this discrepancy in translation from bench science to clinic?
7.1 Limitations of Electrophysiologic Tools
Unlike Electrocardiograms (ECG/EKG), which are standard of care in heart attack evaluations; stroke, with its similarities of ischemia, rarely receives evaluation by an electroencephalogram (EEG) until the following day, if at all. EEG gives an electrical reflection of the brain’s activity. In focal epilepsy, such as TLE, that can be focal waves or spikes [60]. In addition, electrical abnormalities consistent with encephalopathy are often seen in stroke patients when EEG is performed. Yet, many rural hospitals do not utilize EEG to monitor patients with these neurologic conditions because of limitations in skilled personnel and of the equipment itself. Whether evaluation is in a mouse model or a hospital, the spatial discernment sacrificed by electroencephalography due to the bony scalp and other confounding obstacles between the electrodes and the signal cannot always be overcome by the timing integrity of the machine. Event related potentials, frequency spectrum analysis, and other mechanisms of analysis assist in overcoming these limitiations [15]. However, training and validation of the researcher’s technique can consume large amounts of research time.
7.2 Overcoming EEG limitations
Microelectrode arrays and local field potentials show promise in the prediction and localization of pre-ictal activity prior to seizures in TLE [61]. As these tools develop new limitations emerge. For example, a micogrid covers a large surface area in a mouse, but in a human in is a very small area for analysis. This is where the growth of molecular biology and electropyhsiology targets the analysis of these small areas with the precision of basic science. With these developing technologies, the opportunity for molecular biology to create biomarkers of electrophysiologic dysfunction in diseases such as TLE, stroke, and TBI is present and could prove to be the translation required to bring bench science more effectively to the clinic.
7.3 Pharmaceutical Limitations
Anticonvulsant pharmaceutical products create the first line of defense for clinicians treating patients with epilepsy. However, many of the medications are non-linear in their kinetics which means their half-life and clearance are dependent upon plasma concentrations which can fluctuate, reducing the ability to maintain steady state of the drug. In addition, anticonvulsants such as Valproic acid, and Clonazepam are greater than 88% protein bound so they do not cross the blood brain barrier well and therefore provide little if any medication to the targeted areas. In addition, Valproic acid and similar drugs demonstrate serum protein saturable binding which can impact other medications when given simultaneously [62]. Newer anticonvulsants such as ethosuximide and topirimate are more promising, yet the disease is far from controlled. Stroke pharmaceutical options are more limited in that after decades of research we have essentially one medication; tissue plasminogen activator (tPA) that can be used in stroke and that is limited to specific time frames, and patient types. Finally, in TBI there are multiple complicating factors with medication management due to the complexity of symptoms from post-traumatic stress disorder to epilepsy.
Therefore, improved target evaluation and treatment based on a demonstrated abnormality of electrophysiologic dysfunction offers a significant opportunity to identify biomarkers for bioengineering and innovative technologies for multiple neurologic diseases.
7.4 Ion Channel Options
Ion channel targeting offers unchartered opportunity for further evaluation and treatment for TLE, stroke and TBI. Research demonstrates the adaptation of neurons across cellular types[63]. The integration of channel structure and function with cellular stability is described and accepted across research fields, though all the intricacies of how that works are not yet understood. Continuing the process of collaborative research evaluating the interactions and correlations of multiple variables from genetics to inflammatory response by precise research of atomic chemistry at the ion channel level can expose those unknowns at the molecular level. In addition, the collaboration of scientists and technology can compile research data that can be integrated at unprecedented speeds and with increased safety. The Department of Defense, National Institutes of Health (NIH), and other large organizations recognize the increasing need for better outcomes for patients suffering with neurologic diseases like TBI, stroke, and TLE. NIH published a notice in the NIH guide in 2011 encouraging the scientific community to address the issues of predictive failure in preclinical research [64]. The focus to bring basic science to the bedside more efficiently, safely, and effectively creates an opportunity for research that can begin at this primary level and build forward. Evaluating the process of interaction between hyperpolarized HCN1 channels with its unique polarity, depolarized calcium channels, and NKCC1/ KCC2 ratio shifts in neuropathology creates a single variable of interaction that can be modeled in a multi-scale modeling system with a designed control of homeostasis juxtaposed to the actual cellular expression identified by genetic analysis of diseased human tissue. The single constant of a homeostatic model that is computerized and easily modified as new data arrives lends itself not only to comparison against disease tissue, but also better understanding of homeostasis as we start to define the variation within normal. It is against this contrast that we will bridge the gap from bench to bedside and create - in real time - questions that can be answered safely and with better precision before heading to animal and clinical labs for experimentation. This can reduce the time to successful treatment in clinical application and increase efficacy and safety as we identify potential mechanisms that lead to neurodegenerative changes. TLE hippocampal sclerosis, ischemia in stroke, and blood brain barrier compromise in TBI can establish a baseline evaluation of electrophysiologic dysfunction. Then interaction and interdependence of the HCN1 channel, CaV channel and the NKCC1/KCC2 co-transporter channel can correlate dysfunction with structure.
8. Future Research Direction
8.1 Building a Mouse Model
Location/Vascular: A global stroke model in mice creates hippocampal ischemia by way of the posterior circulation consistent with cardiac arrest as well as stroke. Mice have more collateral vascular systems unlike the end artery system in humans and the divergence of humans from that evolutionary past may hold one of the keys to improved biomarker targeting for humans. Therefore, comparison of the mouse model hippocampal data to human supplemental data can expose mechanisms for translational failures of mouse stroke models to clinical application and open doors for other neurologic disease discussion such as epilepsy and TBI as discussed below.
Morphology: Pyramidal neurons and astrocyte immunohistochemistry evaluation within the hippocampus can be used to demonstrate where - on the cellular structures - inclusion of HCN1 channels in control, ischemic injury mice, and hemorrhagic injury mice occurs. Further immunohistochemistry evaluation of mice from control, ischemia, and hemorrhagic stroke groups, triggered to seizure with kainic acid, can be analyzed for HCN1 channel expression prior to and after seizure. This further characterizes location and identifies spatial relationships for comparison to later human research. HCN1 channels with their extracellular exposed post-translational modification tail may demonstrate significant changes and prove a key biomarker to stabilize the first coordinate of a tricoordinate system.
Structure: mRNA sequencing and Western blot for protein expression of HCN1 will reveal increased or decreased expression changes in hippocampal regions exposed to specific injury.
Function: Acute fresh tissue for electrophysiologic evaluation with specific voltage data by patch clamp on dissected hippocampal tissue can then deliver functional data to correlate with specific structure, morphologic, and anatomical data. This precision analysis creates the foundation for a model of translational human tissue analysis.
9. Supplemental Research
9.1 Human Research
Electrophysiology State of the Art Technique: IRB approved human research that can safely evaluate human tissue electrophysiologicaly from initial injury (EEG), through craniotomy and intraoperative electrophysiologic monitoring (intraoperative EEG) during tissue resection. Discarded tissue can then be evaluated for its voltage and current by patch clamp analysis to specify actual electrophysiologic function for comparison to macro tool evaluation. That electrophysiologic data can then be connected with the structural expression of biomarker channels demonstrated in spatial localization by immunohistochemistry and total expression of mRNA and protein by RT-PCR and western blot.
9.2 Building the Future
Multi-scale modeling: Data programmed from the evaluation of the HCN1 hyperpolarizing channel, and additional channels such as the depolarizing calcium channels, and the flow through NKCC1/KCC2 co-transporter channels to demonstrate what is seen experimentally. This programmable data can then be used in simulations of tri-coordinate biomarkers for targeted cell therapy as a translational tool for future experiments prior to clinical application trials.
Acknowledgments
Dr. Nelson Spruston, Dr. Mark Olfert, and Dr. Bernard Schreurs for their kindness and early encouragement in this work, Dominic Quintana for the unique casting of the mouse brain vasculature for figure one, and Penny Hoggarth for typing review and recommendations. Thank you to West Virginia Clinical and Translational Science Institute (WVCTSI), Southern Regional Education Board (SREB), and National Institutes of Health (NIH) for resources and support. Brandon Lucke-Wold received pre-doctoral fellowships from the American Association of Pharmaceutical Scientists and American Foundation of Pharmaceutical Education.
Author Contributions
Gina Sizemore contributed original manuscript concept and writing. Brandon Lucke-Wolde contributed structure edits and neurotrauma recommendations. Charles Rosen, Sanjay Bhatia, James Simpkins, and Dandan Sun contributed valuable recommendations, discussions, and edits in neuroanatomy, neuroinflammation, and ion electrophysiology.
Funding
None.
Competing Interests
The authors have declared that no competing interests exist.
References
- Kobow K, Blümcke I. Epigenetics in epilepsy. Neurosci Lett. 2018; 667: 40-46. [CrossRef] [Google scholar] [PubMed]
- Karhunen H, Bezvenyuk Z, Nissinen J, Sivenius J, Jolkkonen J, Pitkänen A. Epileptogenesis after cortical photothrombotic brain lesion in rats. Neuroscience. 2007; 148: 314-324. [CrossRef] [Google scholar] [PubMed]
- Menon B, Shorvon SD. Ischaemic stroke in adults and epilepsy. Epilepsy Res. 2009; 87: 1. [CrossRef] [Google scholar] [PubMed]
- Khosravani H, Zamponi GW. Voltage-gated calcium channels and idiopathic generalized epilepsies. Physiol Rev. 2006; 86: 941-966. [CrossRef] [Google scholar] [PubMed]
- Orsini A, Zara F, Striano P. Recent advances in epilepsy genetics. Neurosci Lett. 2018; 667: 4-9. [CrossRef] [Google scholar] [PubMed]
- Vahedi K, Depienne C, Le FD, Riant F, Chaine P, Trouillard O, et al. Elicited repetitive daily blindness: a new phenotype associated with hemiplegic migraine and SCN1A mutations. Neurology. 2009; 72: 1178. [CrossRef] [Google scholar] [PubMed]
- Pellock JM, Arzimanoglou A, D'Cruz ON, Holmes GL, Nordli D, Shinnar S. Extrapolating evidence of antiepileptic drug efficacy in adults to children ≥2years of age with focal seizures: The case for disease similarity. Epilepsia. 2017; 73: 90-94. [CrossRef] [Google scholar] [PubMed]
- Kucukyuruk B, Richardson RM, Wen HT, Fernandezmiranda JC, Jr RA. Microsurgical anatomy of the temporal lobe and its implications on temporal lobe epilepsy surgery. Epilepsy Res Treat. 2012; 2012: 769825. [CrossRef] [Google scholar] [PubMed]
- Nyquist P, Bautista C, Jichici D, Burns J, Chhangani S, Defilippis M, et al. Prophylaxis of venous thrombosis in neurocritical care patients: An evidence-based guideline: A statement for healthcare professionals from the neurocritical care society. Crit Care Med. 2017; 45: 476. [CrossRef] [Google scholar] [PubMed]
- Mckee AC, Daneshvar DH. The neuropathology of traumatic brain injury. Handb Clin Neurol. 2015; 127: 45-66. [CrossRef] [Google scholar] [PubMed]
- Zhang JH. Vascular neural network in subarachnoid hemorrhage. Transl Strok Res. 2014; 5: 423-428. [CrossRef] [Google scholar] [PubMed]
- Marhold F, Rosen CL. Novel technique to improve vessel mismatch when using saphenous vein bypass grafts for intracranial revascularization procedures. J Neurosurg. 2010; 112: 1227-1231. [CrossRef] [Google scholar] [PubMed]
- Turner RC, Dodson SC, Rosen CL, Huber JD. The science of cerebral ischemia and the quest for neuroprotection: navigating past failure to future success. J Neurosurg. 2013; 118: 1072-1085. [CrossRef] [Google scholar]
- Ward NS. Does neuroimaging help to deliver better recovery of movement after stroke? Curr Opin Neurol. 2015; 28: 323. [CrossRef] [Google scholar]
- Iyer KK. Effective assessments of electroencephalography during stroke recovery: contemporary approaches and considerations. J Neurophysiol. 2017;118: 2521-2525. [CrossRef] [Google scholar] [PubMed]
- Pivonkova H, Anderova M. Altered homeostatic functions in reactive astrocytes and their potential as a therapeutic target after brain ischemic injury. Curr Pharm Des. 2017; 23: 5056-5074. [CrossRef] [Google scholar] [PubMed]
- Troakes C, Smyth R, Noor F, Maekawa S, Killick R, King A, et al. Clusterin expression is upregulated following acute head injury and localizes to astrocytes in old head injury. Neuropathology. 2016; 37: 12-24. [CrossRef] [Google scholar] [PubMed]
- Albrecht J, Zielińska M. Mechanisms of Excessive Extracellular Glutamate Accumulation in Temporal Lobe Epilepsy. Neurochem Res. 2016; 42: 1724-1734. [CrossRef] [Google scholar] [PubMed]
- Cembrowski MS, Spruston N. Integrating Results across Methodologies Is Essential for Producing Robust Neuronal Taxonomies. Neuron. 2017; 94: 747. [CrossRef] [Google scholar] [PubMed]
- Quintana P, Soto D, Poirot O, Zonouzi M, Kellenberger S, Muller D, et al. Acid‐sensing ion channel 1a drives AMPA receptor plasticity following ischaemia and acidosis in hippocampal CA1 neurons. J Physiol. 2015; 593: 4373-4386. [CrossRef] [Google scholar] [PubMed]
- Spruston N. Pyramidal neurons: dendritic structure and synaptic integration. Nat Rev Neurosci. 2008; 9: 206. [CrossRef] [Google scholar] [PubMed]
- Bhuiyan MI, Song S, Yuan H, Begum G, Kofler J, Kahle KT, et al. WNK-Cab39-NKCC1 signaling increases the susceptibility to ischemic brain damage in hypertensive rats. J Cereb Blood Flow Metab. 2016; 37: 2780-2794. [CrossRef] [Google scholar] [PubMed]
- Muradashvili N, Tyagi SC, Lominadze D. Localization of fibrinogen in the vasculo-astrocyte interface after cortical contusion injury in mice. Brain Sci. 2017; 7. [CrossRef] [Google scholar] [PubMed]
- Brennan GP, Henshall DC. microRNAs in the pathophysiology of epilepsy. Neurosci Lett. 2018; 667: 47-52. [CrossRef] [Google scholar] [PubMed]
- Weiss N, Miller F, Cazaubon S, Couraud PO. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta. 2009; 1788: 842-857. [CrossRef] [Google scholar] [PubMed]
- Bukeirat M, Sarkar SN, Hu H, Quintana DD, Simpkins JW, Ren X. MiR-34a regulates blood-brain barrier permeability and mitochondrial function by targeting cytochrome c. J Cereb Blood Flow Metab. 2016; 36: 387. [CrossRef] [Google scholar] [PubMed]
- Hasbargen T, Ahmed MM, Miranpuri G, Li L, Kahle KT, Resnick D, et al. Role of NKCC1 and KCC2 in the development of chronic neuropathic pain following spinal cord injury. Anna N Y Acad Sci. 2010; 1198: 168–172. [CrossRef] [Google scholar] [PubMed]
- Lee KPK, Chen J, MacKinnon R. Molecular structure of human KATP in complex with ATP and ADP. Elife. 2017; 6. [CrossRef] [Google scholar] [PubMed]
- O'Connor ER, Sontheimer H, Spencer DD, de Lanerolle NC. Astrocytes from human hippocampal epileptogenic foci exhibit action potential-like responses. Epilepsia. 1998; 39: 347. [CrossRef] [Google scholar] [PubMed]
- Jaggi AS, Kaur A, Bali A, Singh N. Expanding spectrum of sodium potassium chloride co-transporters in the pathophysiology of diseases. Curr Neuropharmacol. 2015; 13: 369-388. [CrossRef] [Google scholar] [PubMed]
- Cong D, Zhu W, Kuo JS, Hu S, Sun D. Ion transporters in brain tumors. Curr Med Chem. 2015; 22: 1171-1181. [CrossRef] [Google scholar]
- Adragna NC, Ravilla NB, Lauf PK, Begum G, Khanna AR, Sun D, et al. Regulated phosphorylation of the K-Cl cotransporter KCC3 is a molecular switch of intracellular potassium content and cell volume homeostasis. Front Cell Neurosci. 2015; 9: 255. [CrossRef] [Google scholar]
- Biel M, Wahlschott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: From genes to function. Physiol Rev. 2009; 89: 847-885. [CrossRef] [Google scholar] [PubMed]
- Benari Y, Khalilov I, Kahle KT, Cherubini E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist. 2012; 18: 467-486. [CrossRef] [Google scholar] [PubMed]
- Brennan GP, Baram TZ, Poolos NP. Hyperpolarization-Activated cyclic nucleotide-gated (HCN) channels in epilepsy. Cold Spring Harb Perspect Med. 2016; 6: a022384. [CrossRef] [Google scholar] [PubMed]
- DiFrancesco JC, DiFrancesco D. Dysfunctional HCN ion channels in neurological diseases. Front Cell Neurosci. 2015; 6: 174. [CrossRef] [Google scholar] [PubMed]
- Lee CH, Mackinnon R. Structures of the human HCN1 hyperpolarization-activated channel. Cell. 2017; 168: 111-120. [CrossRef] [Google scholar] [PubMed]
- Luckewold BP, Turner RC, Logsdon AF, Bailes JE, Huber JD, Rosen CL. Linking traumatic brain injury to chronic traumatic encephalopathy: identification of potential mechanisms leading to neurofibrillary tangle development. J Neurotrauma. 2014; 31: 1129-1138. [CrossRef] [Google scholar] [PubMed]
- Weerasinghe D, Menon P, Vucic S. Hyperpolarization-activated cyclic-nucleotide-gated channels potentially modulate axonal excitability at different thresholds. J Neurophysiol. 2017; 118: 3044-3050. [CrossRef] [Google scholar] [PubMed]
- Turlova E, Feng ZP, Sun HS. The role of TRPM2 channels in neurons, glial cells and the blood-brain barrier in cerebral ischemia and hypoxia. Acta Pharmacol Sin. 2018; 39: 713-721. [CrossRef] [Google scholar] [PubMed]
- Mannix R, Berkner J, Mei Z, Alcon S, Hashim J, Robinson S, et al. Adolescent mice demonstrate a distinct pattern of injury after repetitive mild traumatic brain injury. J Neurotrauma. 2017; 34: 495-504. [CrossRef] [Google scholar] [PubMed]
- Faria LC, Gu F, Parada I, Barres B, Luo ZD, Prince DA. Epileptiform activity and behavioral arrests in mice overexpressing the calcium channel subunit alpha2delta-1. Neurobiol Dis. 2017; 102: 70-80. [CrossRef] [Google scholar] [PubMed]
- Calejo AI, Reverendo M, Silva VS, Pereira PM, Santos MA, Zorec R, et al. Differences in the expression pattern of HCN isoforms among mammalian tissues: sources and implications. Mol Biol Rep. 2014; 41: 297-307. [CrossRef] [Google scholar] [PubMed]
- Santina LD, Piano I, Cangiano L, Caputo A, Ludwig A, Cervetto L, et al. Processing of retinal signals in normal and HCN deficient mice. PloS One. 2012; 7: e29812. [CrossRef] [Google scholar] [PubMed]
- Kahle KT, Merner ND, Friedel P, Silayeva L, Liang B, Khanna A, et al. Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 2014; 15: 766–774. [CrossRef] [Google scholar] [PubMed]
- Merner ND, Chandler MR, Bourassa C, Liang B, Khanna AR, Dion P, et al. Regulatory domain or CpG site variation in SLC12A5, encoding the chloride transporter KCC2, in human autism and schizophrenia. Front Cell Neurosci. 2015; 9: 386. [CrossRef] [Google scholar] [PubMed]
- PhD KTKM, Kevin Staley MD. Altered Neuronal Chloride Homeostasis and Excitatory GABAergic Signaling in Human Temporal Lobe Epilepsy. Epilepsy Curr. 2008; 8: 51-53. [CrossRef] [Google scholar] [PubMed]
- Saitsu H, Watanabe M, Akita T, Ohba C, Sugai K, Ong WP, et al. Impaired neuronal KCC2 function by biallelic SLC12A5 mutations in migrating focal seizures and severe developmental delay. Sci Rep. 2016; 6: 30072. [CrossRef] [Google scholar] [PubMed]
- Frederikse PH, Kasinathan C. KCC2 expression supersedes NKCC1 in mature fiber cells in mouse and rabbit lenses. Mol Vis. 2015; 21: 1142-1150. [Google scholar]
- Tsukahara T, Masuhara M, Iwai H, Sonomura T, Sato T. Repeated stress-induced expression pattern alterations of the hippocampal chloride transporters KCC2 and NKCC1 associated with behavioral abnormalities in female mice. Biochem Biophys Res Commun. 2015; 465: 145-151. [CrossRef] [Google scholar] [PubMed]
- Li X, Zhou J, Chen Z, Chen S, Zhu F, Zhou L. Long-term expressional changes of Na+ -K+ -Cl- co-transporter 1 (NKCC1) and K+ -Cl- co-transporter 2 (KCC2) in CA1 region of hippocampus following lithium-pilocarpine induced status epilepticus (PISE). Brain Res. 2008; 1221: 141-146. [CrossRef] [Google scholar] [PubMed]
- Gagnon KB, Delpire E. Physiology of SLC12 transporters: lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am J Physiol Cell Physiol. 2013; 304: C693-714. [CrossRef] [Google scholar] [PubMed]
- Tian Y, Guo SX, Li JR, Du HG, Wang CH, Zhang JM, et al. Topiramate attenuates early brain injury following subarachnoid haemorrhage in rats via duplex protection against inflammation and neuronal cell death. Brain Res. 2015; 1622: 174. [CrossRef] [Google scholar] [PubMed]
- Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. 2015; 67: 821. [CrossRef] [Google scholar] [PubMed]
- Hennessey JA, Boczek NJ, Jiang YH, Miller JD, Patrick W, Pfeiffer R, et al. A CACNA1C variant associated with reduced voltage-dependent inactivation, increased CaV1.2 channel window current, and arrhythmogenesis. PloS One. 2014; 9: e106982. [CrossRef] [Google scholar] [PubMed]
- Nietorostro M, Sandhu G, Bauer CS, Jiruska P, Jefferys JGR, Dolphin AC. Altered expression of the voltage-gated calcium channel subunit α2δ-1: A comparison between two experimental models of epilepsy and a sensory nerve ligation model of neuropathic pain. Neuroscience. 2014; 283: 124-137. [CrossRef] [Google scholar] [PubMed]
- Pirone A, Kurt S, Zuccotti A, Rüttiger L, Pilz P, Brown DH, et al. α2δ3 is essential for normal structure and function of auditory nerve synapses and is a novel candidate for auditory processing disorders. J Neurosci. 2014; 34: 434. [CrossRef] [Google scholar] [PubMed]
- Tuluc P, Molenda N, Schlick B, Obermair GJ, Flucher BE, Jurkatrott K. A CaV1.1 Ca2+ channel splice variant with high conductance and voltage-sensitivity alters EC coupling in developing skeletal muscle. Biophys J. 2009; 96: 35-44. [CrossRef] [Google scholar] [PubMed]
- McGonigle P, Ruggeri B. Animal models of human disease: challenges in enabling translation. Biochem Pharmacol. 2014; 87: 162-171. [CrossRef] [Google scholar] [PubMed]
- Stafstrom CE, Carmant L. Seizures and epilepsy: an overview for neuroscientists. Cold Spring Harb Perspect Med. 2015; 5: a022426-a022426. [CrossRef] [Google scholar] [PubMed]
- Aghagolzadeh M, Hochberg LR, Cash SS, Truccolo W, Aghagolzadeh M, Hochberg LR, et al., editors. Predicting seizures from local field potentials recorded via intracortical microelectrode arrays. Conf Proc IEEE Eng Med Biol Soc. 2016: 6353-6356. [CrossRef] [Google scholar] [PubMed]
- Patsalos PN, Zugman M, Lake C, James A, Ratnaraj N, Sander JW. Serum protein binding of 25 antiepileptic drugs in a routine clinical setting: A comparison of free non-protein-bound concentrations. Epilepsia. 2017; 58: 1234-1243. [CrossRef] [Google scholar] [PubMed]
- Wolfart J, Laker D. Homeostasis or channelopathy? Acquired cell type-specific ion channel changes in temporal lobe epilepsy and their antiepileptic potential. Front Physiol. 2015; 6: 168. [CrossRef] [Google scholar] [PubMed]
- Landis SC, Amara SG, Asadullah K, Austin CP, Blumenstein R, Bradley EW, et al. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012; 490: 187-191. [CrossRef] [Google scholar] [PubMed]

