

Distinct Mechanisms of Alterations in DNA Methylation/Demethylation Leading to Myelodysplastic Syndromes/Acute Myeloid Leukemia and Chronic Lymphocytic Leukemia
Cristina Bagacean 1,2,3![]() , Anne Bordron 3
, Anne Bordron 3![]() , Tempescul Adrian 1,3
, Tempescul Adrian 1,3![]() , Jean Christophe Ianotto 3
, Jean Christophe Ianotto 3![]() , Gaelle Guillerm 3
, Gaelle Guillerm 3![]() , Wesley H Brooks 4
, Wesley H Brooks 4![]() , Marie-Anne Couturier 3
, Marie-Anne Couturier 3![]() , Mihnea Zdrenghea 5
, Mihnea Zdrenghea 5![]() , Christian Berthou 1,3
, Christian Berthou 1,3![]() , Yves Renaudineau 2,3,*
, Yves Renaudineau 2,3,* ![]()
- Department of Clinical Hematology, CHU Morvan, Brest, France
- Laboratory of Immunology and Immunotherapy, CHU Morvan, Brest, France
- INSERMU1127, B lymphocyte and autoimmunity, INSERM, University of Brest, REpiCGO, ICCGO, Brest, France
- Department of Chemistry, University of South Florida, Tampa, USA
- Department of Hematology, "Prof. Dr. Ion Chiricuta" Oncology Institute, Cluj-Napoca, Romania
* Correspondence: Yves Renaudineau ![]()
Received: June 05, 2018 | Accepted: November 28, 2018 | Published: December 14, 2018
OBM Genetics 2018, Volume 2, Issue 4 doi: 10.21926/obm.genet.1804054
Academic Editors: Marcel Mannens and Stéphane Viville
Special Issue: Epigenetic Mechanisms in Health and Disease
Recommended citation: Bagacean C, Bordron A, Adrian T, Ianotto JC, Guillerm G, Brooks WH, Couturier MA, Zdrenghea M, Berthou C, Renaudineau Y. Distinct Mechanisms of Alterations in DNA Methylation/Demethylation Leading to Myelodysplastic Syndromes/Acute Myeloid Leukemia and Chronic Lymphocytic Leukemia. OBM Genetics 2018;2(4):054; doi:10.21926/obm.genet.1804054.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Epigenetic dysregulation is present in both myeloid and lymphoid disorders, with important differences reported between myelodysplastic syndromes (MDS)/acute myeloid leukemia (AML), on one hand, and chronic lymphocytic leukemia (CLL), on the other. Qualitative differences are reported in MDS/AML with gene fusions (e.g. TET1/LCX) and somatic mutations in epigenetic regulators (e.g. DNMT3A, TET2, IDH1/2), while differences in CLL are predominantly quantitative (e.g. DNMT3A, TET2). Indeed, and as supported by studies in animal models, a defective DNA methylation/demethylation process represents a competitive advantage to the myeloid lineage and an early event in MDS/AML, while in the case of CLL, epigenetic events appear later and are associated with disease progression. Finally, in both MDS/AML and CLL, the focal or global DNA methylation/demethylation process is altered and contributes to disease progression and activity. In conclusion, a better understanding of the epigenetic regulators involved in myeloid/lymphoid differentiation, their localization and the co-recruitment of other proteins at specific DNA target sites, could offer us the possibility to modulate hematopoiesis, and control disease initiation and/or progression.
Keywords
DNA methylation; DNA hydroxymethylation; MDS; AML; CLL; TETs; DNMTs
1. Introduction
Myelodysplastic syndromes (MDS) comprise a diverse group of clonal and malignant myeloid disorders characterized by ineffective hematopoiesis, peripheral cytopenias, and an increased risk of progression to acute myeloid leukemia (AML), either gradually or rapidly [1]. AML is the most frequently occurring acute leukemia in adults, with malignant transformation occurring in primitive hematopoietic stem and progenitor cells, leading to increased proliferation and impaired differentiation [2,3]. A wide heterogeneity of genetic mutations has been identified in both MDS and AML, the most frequent involving epigenetic processes, including the control of DNA methylation, RNA splicing, histone modifications, transcription factor function, DNA repair, and kinase signaling.
Another hematologic pathology presenting important epigenetic modifications, is chronic lymphocytic leukemia (CLL), which is the most common type of leukemia, with a median age at diagnosis ranging from 67 to 72 years [4,5]. CLL is characterized by the clonal proliferation and accumulation of mature, long-lived CD5+ B cells in peripheral blood, bone marrow, lymph nodes and secondary lymphoid organs [6]. Several hypotheses have been formulated regarding the cell of origin for CLL B cells. Initially, based on the subdivision of CLL patients into two main subgroups according to their immunoglobulin heavy chain variable region (IGHV) mutational status, it was suspected that CLL cells were derived either from naïve B cells, in the case of the IGHV unmutated (UM) CLL B cells, or from memory B cells, in the case of mutated (M) CLL B cells. Subsequently, this dichotomy lost support with the development of gene expression microarray analysis, which is considered to be the gold standard technique for classifying the different lymphoid malignancies based on their cell of origin. Indeed, according to Klein et al. and validated since, transcriptome analysis revealed only a small number of differences between IGHV-UM and IGHV-M CLL B cells, supporting the possibility that both subsets are related to memory B cells [7]. A recent study has confirmed these findings by integrating DNA methylation data and comparing normal B cell subsets to malignant B cells from 268 patients with CLL. This analysis showed that CLLs are derived largely from a continuum of memory B cell maturation states ranging from low- to high-maturity [8] In addition, such analysis verified the observation that the CLL methylation status resembles the that of memory B cells, accounting for 70%–100% of the programming for high-maturity memory B cells. However, the debate is not closed regarding the origin of CLL cells, and there is mounting evidence supporting the occurrence of the primary oncogenic event not at the memory B cell stage, but earlier in the HSC counterpart, and that the leukemogenic transformation is a step-wise process involving the accumulation of oncogenic events during B cell differentiation to the memory B cell stage [9]. In MDS/AML, somatic mutations are presented in epigenetic regulators. In contrast, in CLL, epigenetic modifications are not qualitative, but mainly quantitative and affect the expression of epigenetic sensitive genes, such as CD5 and CD20, with differences reported during CLL disease progression [10,11,12]. Such a dichotomy may explain the absence of the simultaneous occurrence of CLL and MDS/AML, except in CLL patients receiving chemotherapeutic and mutagenic agents.
Accordingly, the aim of this review is to discuss the parallels between MDS/AML and CLL, diseases that represent two different mechanisms by which the epigenetic landscape is modified: by somatic mutations (qualitative) in MDS/AML, and at the expression level (quantitative) in CLL. Concerning MDS/AML, epigenetic dysregulation is an early event, conferring a competitive advantage on the myeloid lineage and leading to self-renewal. On the other hand, in CLL, there is a step-wise process in which methylation dysregulation represents a secondary event and contributes to disease progression.
2. DNA Methylation and Demethylation
DNA methylation occurs predominantly at position 5 of the cytosine and represents the most studied epigenetic marker. Research on this cytosine methylation in cancer began in the 1970s when analysis of tumors revealed significant aberrations in 5-methylcytosine (5-mCyt). Furthermore, in 2009, rapid breakthroughs came with the characterization of an active DNA demethylation enzymatic process that starts with the conversion of 5-mCyt to 5-hydroxy(h)mCyt (Figure 1) [13,14].
Currently, due to important advances in DNA methylation analysis technologies, whole-genome scale methylation maps are available for several human cell types and show that the methylation is mainly restricted to cytosine-phospho-guanosine (CpG) sites. The human genome contains approximately 29 million CpG sites, 60% to 80% of which are generally methylated and with important differences among cell types [15]. Compartment-specific analysis of the methylome showed that DNA methylation has an important role in maintaining the latent transcriptional repression of pericentromeric repeats and transposable repetitive elements [16,17,18,19]. This is essential for genomic stability and normal mitosis, by controlling chromosome alignment, segregation and integrity. The human genome is globally methylated with the exception of a small fraction (1%–2%) of the total genome which possesses CpG rich spots, referred to as CpG islands. These regions are predominantly located in transcriptionally active gene areas and are usually close to the first exon of a gene. The asymmetric distribution of DNA methylation between cell types has been shown to have functional implications in the epigenetic guidance of development, cellular differentiation and more recently, in nuclear organization; however, our understanding of this distribution and its effects is still incomplete [20,21,22,23]. Approximately 70% of gene promoters overlap with CpG islands and it is generally accepted that the state of CpG methylation in these DNA regions, regulates gene expression through modification of chromatin structure and accessibility to the transcription machinery [24,25]. The methylation process is catalyzed by the DNA methyltransferase (DNMT) enzymes, including DNMT1, DNMT3A, and DNMT3B, with the help of accessory proteins, such as DNMT3L and UHRF1.
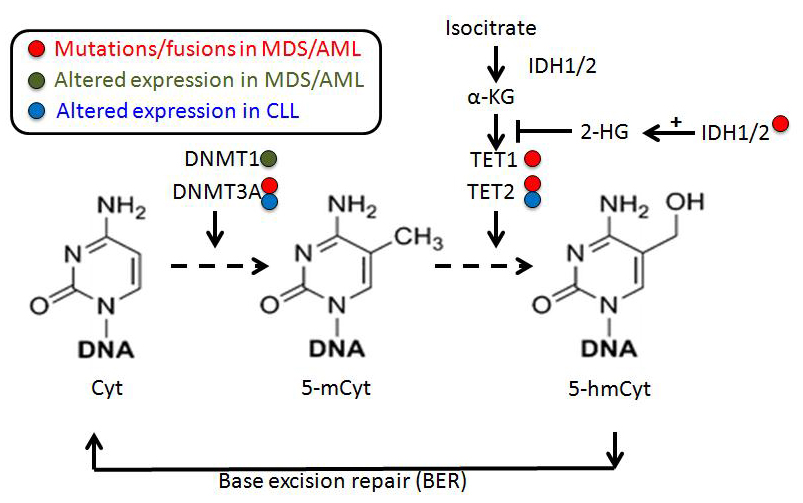
Figure 1 Pathways of cytosine methylation and demethylation mediated by DNA methyltransferase (DNMT) and ten-eleven translocation (TET) proteins.
(Note: In MDS/AML, the main DNA methylation/demethylation regulators are DNMT1 (overexpressed), DNMT3A and TET2, presenting loss of function due to mutations, and IDH1/2 that repress TET2 through α-KG inhibition when mutated. In CLL, DNMT3A and TET2 expression is altered during disease progression. Cyt: cytosine; 5-mCyt: 5-methylcytosine; 5-hmCyt: 5-hydroxymethylcytosine; α-KG: α-ketoglutarate; 2-HG: 2-hydroxy-glutarate; IDH1/2: isocitrate dehydrogenase. MDS: myelodysplastic syndrome; AML: acute myeloid leukemia; CLL: chronic lymphocytic leukemia.)
DNMTs catalyze the transfer of a methyl group from the methyl donor S-adenosyl-L-methionine (SAM), to position 5 of cytosine residues in DNA [26]. SAM, in turn, is consumed by increased activation of the polyamine synthesis and recycling pathways [27]. The first DNMT to be purified was shown to have a preferential affinity for hemi-methylated DNA substrates, and therefore is considered responsible for maintaining the methylation pattern during DNA replication [28]. The gene encoding this enzyme was subsequently cloned and sequenced by the same group and named DNMT1 [29].
The observation that DNA methylation is not completely lost after DNMT1 inactivation suggested that cells must contain a de novo methylation activity. Later, Li and colleagues identified and cloned two novel methyltransferases [30]. These genes are expressed as two proteins, DNMT3A and DNMT3B, with similar structure and domain organization. These enzymes do not differentiate between hemi-methylated and unmethylated CpG sites and are responsible for de novo DNA methylation, in association with DNMT3L, which has a regulatory function and no methylation capacity per se [31].
Regarding the active DNA demethylation process, 5-hmCyt was identified shortly after the discovery of 5-mCyt [32]. After this discovery, no remarkable research was published on the subject, until the ten-eleven translocation (TET)1 protein was discovered and shown to be capable of erasing methylation marks via oxidation to 5-hmCyt [14]. By definition, active DNA demethylation refers to the enzymatic process by which 5-mCyt and demethylation intermediates are replaced by an unmodified cytosine in a replication-independent manner [33]. In contrast, the failure to maintain DNA methylation marks across cell divisions, resulting in the dilution of 5-mCyt during replication, is referred to as passive DNA demethylation. TET1 belongs to a family of enzymes which also includes TET2 and TET3 and catalyze the successive transformation of 5-mCyt to 5-hmCyt, 5-formylcytosine (5-fCyt) and 5-carboxylcytosine (5-CaCyt) in a Fe(II)- and 2-oxoglutarate-dependent manner [34,35].
Methylation genome mapping provided important insights into the role of 5-mCyt. Similarly, genome mapping of the demethylation process intermediates, and in particular 5-hmCyt, have been shown to be associated with important epigenetic regulatory functions. Indeed, 5-hmCyt content is highly variable in normal human tissues and higher levels of 5-hmCyt content are found in areas of transcriptional control, including promoters, transcription factor binding sites and gene bodies [36]. However, as highlighted in a recently published study, the relationship between 5-hmCyt peaks and gene transcription is complex, which raises important questions and challenges based on some of the data provided so far by DNA methylome analysis using microarrays or the reduced representation bisulfite sequencing (RRBS) techniques [37]. In this study, Zhang et al. demonstrated that the 5-mCyt distribution is only poorly correlated with gene expression, whereas the 5-hmCyt displays a “bi-modal” pattern in HSC with a high 5-hmCyt distribution in the promoter and gene body associated with highly expressed genes, while high 5-hmCyt levels in the transcription start sites (TSS) reveal lower relative gene expression [37]. The positive association between high 5-hmCyt content in the gene bodies with gene expression in embryonic HSC was also demonstrated in a study by Huang et al. which, additionally, demonstrated a correlation with TET2 activity [38].
Another interesting observation concerning the mechanisms by which the fragile balance between methylation and hydroxymethylation is adjusted is that DNMT3A and TET1 function in a complementary and competitive manner, as demonstrated by Gu et al [39]. This study showed that normal epigenetic landscapes and gene expression are maintained by TET1, which limits DNA methylation partially by protecting the cytosine target from DNMT3A and establishing boundaries for DNA methylation.
Methylation and demethylation enzymes have also been shown to contribute to the adjustment of the epigenetic landscape and transcriptional regulation in an enzyme-independent manner, by simply recruiting other enzymes and co-factors. For example, there is a direct interaction of TET2 and TET3 with O‐GlcNAc transferase (OGT) [40]. TET2/3–OGT co‐localizes on chromatin at active promoters enriched for H3K4me3 and reduction of either TET2/3 or OGT activity results in a direct decrease in H3K4me3 and concomitantly decreased transcription. Independent of its catalytic activity, TET1 also has a role in transcriptional repression through its interaction with the SIN3A complex [41]. These interactions complicate the interpretation of the link between the localization on the genome of methylation/hydroxymethylation enzymes, methylated/ hydroxymethylated CpGs and transcriptional regulation.
3. Knowledge Gained from Mouse Models: The Link between DNA Methylation, Myeloid or the Lymphoid Lineage Commitment, Self-Renewal and Differentiation
The hematopoietic system and its hierarchical cascade of differentiation provides comprehensive information on the DNA methylation/demethylation process during differentiation (Figure 2). Indeed, important DNA methylation/demethylation maps have been shown to reflect cell types and differentiation stages [42].
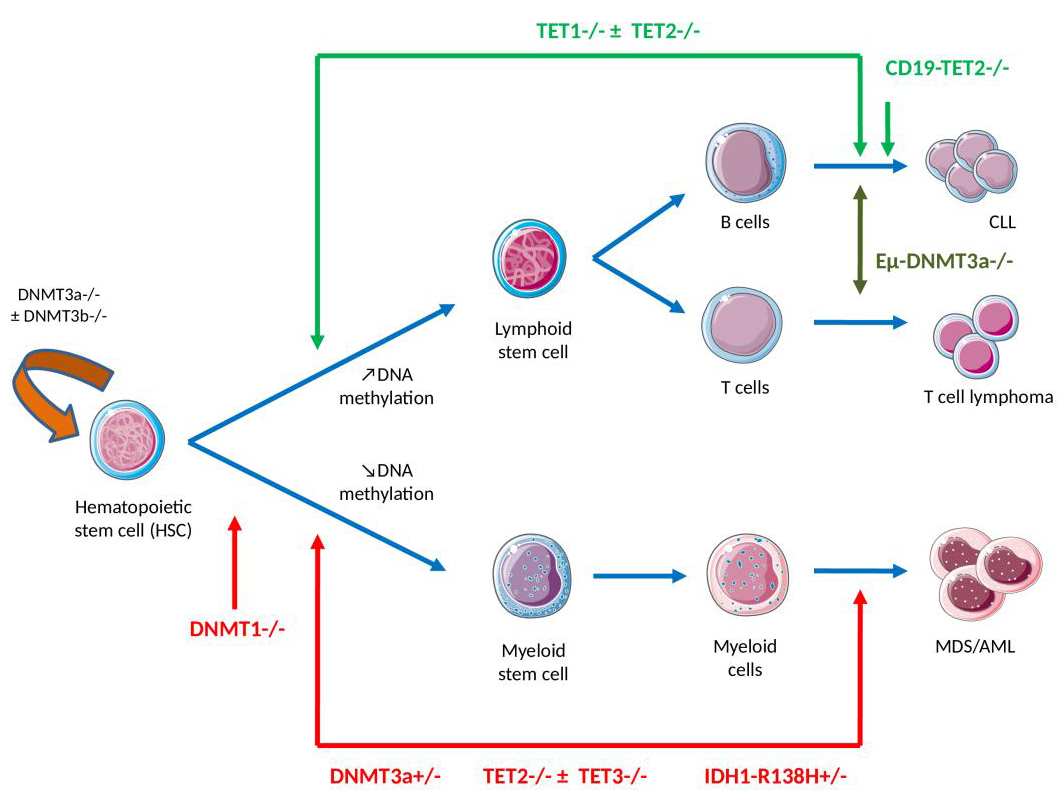
Figure 2 The link between DNA methylation, myeloid or the lymphoid lineage commitment, self-renewal and differentiation – knowledge gained from mouse models. (Note: Lymphoid/myeloid differentiation in the bone marrow compartment is tightly regulated at the epigenetic level, with an observed increase in DNA methylation levels following lymphoid commitment, predominating at myeloid transcription factor binding sites, and a decrease as myeloid differentiation progresses.
DNMT1 inhibition in hematopoietic progenitors skews progenitors toward the myeloid state, causing enhanced cell cycling of myeloid progenitor cells and reduced lymphoid differentiation potential. Homozygous DNMT3A and DNMT3B deficiency at the hematopoietic stem cell (HSC) stage of differentiation generates global DNA demethylation associated with HSC multipotency, self-renewal gene upregulation and downregulation of differentiation factors, which results in a severe block in myeloid/lymphoid differentiation and enhanced HSC self-renewal. Heterozygous Dnmt3a null (Dnmt3a+/–) mice develop myeloid skewing over time and myeloid malignancies after a long latent period.
TET1-deficient mice develop a lymphoproliferative disease at an advanced age (approximately 2 years), with a mature germinal center B cell phenotype. TET2-deficient mice present an altered cell differentiation with skewing toward monocytic/granulocytic lineages observed and development of chronic myelomonocytic leukemia (CMML), myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). The B cell-specific TET2-knockout, on the other hand, leads to abnormalities in the B1 cell subset and a development of B cell malignancies. The combined loss of TET1/TET2 in HSC generates the development of lethal B cell neoplasms whereas the dual loss of TET2/TET3 results in aggressive myeloid leukemia, with an almost complete loss of 5-hmCyt.
IDH1-R132H knock-in mice shared similar phenotypes with the Tet2-knockout mice, including global 5-hmCyt reduction, altered DNA methylation, impaired hematopoietic differentiation, myeloid skewing and the development of myeloid disorders. IDH2-R140Q transgenic mice showed normal HSC numbers and lineage differentiation, but exhibited increased extramedullary hematopoiesis and, when combined with HoxA9/Meis1 or FLT3 mutations in hematopoietic cells, produced acute myeloid leukemia.)
Lymphoid/myeloid differentiation in the bone marrow compartment is tightly regulated at the epigenetic level, with an observed increase in DNA methylation levels following lymphoid commitment, and a decrease as myeloid differentiation progresses [43]. DNA methylation mapping at the CpG level in the primitive hematopoietic compartment has revealed a progressive and specific methylation accumulation during lymphoid differentiation that predominates at myeloid transcription factor binding sites [42]. This suggests that DNA methylation is important for cell fate determination and the balanced commitment to the myeloid or the lymphoid lineages.
Multiple transgenic mouse models have been established to study the role of epigenetics in the hematopoietic lymphoid/myeloid differentiation process. The fact that DNA methylation profiling of murine hematopoietic progenitors involves more global methylation enrichment during early lymphopoiesis than during myelopoiesis could explain why DNMT1 inhibition skews progenitors toward the myeloid state [43]. In other words, these data suggest that DNMT1 differentially controls the myeloid and lymphoid lineages, causing enhanced cell cycling of myeloid progenitor cells and reduced lymphoid differentiation potential [43].
Next, to determine whether constitutive methylation is continuously required throughout lymphoid differentiation, DNMT1 was also inhibited in lymphoid progenitors. In contrast to the previous mouse model, normal numbers and undisturbed homeostasis of B lymphoid cells were detected. Therefore, once the B cell program is established, a lower sustained constitutive DNA methylation is sufficient to maintain B cell identity and maturation [43].
Although it has been clearly shown that lymphoid commitment of the HSC is protected by DNMT1 and involves a global increase in DNA methylation, the roles of DNMT3A and DNMT3B at this early stage of B cell differentiation are less clear. DNMT3A-null HSCs exhibit global DNA demethylation but show both increased and decreased DNA methylation at different loci [44]. These modifications are associated with upregulation of HSC multipotency and self-renewal genes as well as downregulation of differentiation factors. Serial transplantation of DNMT3A-null HSC progressively impairs HSC differentiation [44]. Conditional loss of DNMT3B in HSC, another de novo methyltransferase, results in minimal phenotypic effects; however, combined loss of DNMT3B and DNMT3A results in a more severe block in myeloid/lymphoid differentiation and an enhanced HSC self-renewal similar to that observed in DNMT3A-null HSCs [45] Together, these data suggest a crucial role of de novo methyltransferases, particularly DNMT3A, in determining HSC differentiation and self-renewal, their expression being required to allow HSC to differentiate into the myeloid/lymphoid lineage.
Hematopoiesis was also evaluated in mice heterozygous for a constitutive Dnmt3a null mutation. With no other manipulations, Dnmt3a+/– mice developed myeloid skewing over time and their HSCs exhibited a long-term competitive transplantation advantage. Furthermore, Dnmt3a+/– mice also spontaneously developed transplantable myeloid malignancies after a long latent period [46].
Interestingly, in a different mouse model bearing a defective DNMT3a gene under control of the immunoglobulin heavy chain enhancer (Eμ-DNMT3a-/-), DNMT3A inactivation occurred in both embryonic and adult stem and progenitor hematopoietic cells. Moreover, mice did not develop a myeloid disease but, in contrast, a lymphoproliferative disease resembling CLL or a peripheral T cell lymphoma phenotype [47].
DNA hydroxymethylation enzymes also play important roles in HSC differentiation. In transgenic TET1 knockout mice, HSC analysis showed global loss of 5-hmCyt with greater losses in 5-hmCyt occurring in gene bodies (exons and introns), important gains in 5-mCyt at promoters, and increased self-renewal potential [48]. TET1-deficient mice further developed a lymphoproliferative disease at an advanced age (approximately 2 years), with a mature germinal center (GC) B cell phenotype [48]. In Tet2-deficient mice, an altered cell differentiation skewed toward monocytic/granulocytic lineages was observed and Tet2-knockout mice develop chronic myelomonocytic leukemia (CMML), MDS and AML with increased proliferation of the HSC compartment [49]. It is important to note that the B cell-specific Tet2-knockout (CD19-TET2-/-), on the other hand, leads to abnormalities in the B1 cell subset and a development of B cell malignancies showing similarities to human CLL, including CD5 expression and sensitivity to ibrutinib-mediated B cell receptor (BCR) signaling inhibition [50]. Interestingly, the combined loss of Tet1/Tet2 in HSC is associated with the development of lethal B cell neoplasms, while the dual loss of Tet2/Tet3 results in aggressive myeloid leukemia with an almost complete loss of 5-hmCyt [51,52].
Heterozygous mutations in the catalytic arginine residues of isocitrate dehydrogenases 1 and 2 (IDH1 and IDH2) are common in MDS and LAM and the mutant enzymes acquire a neomorphic activity by converting the TET substrate α-ketoglutarate (α-KG) to D-2-hydroxyglutarate (2-HG). The IDH1-R132H knock-in mice shared similar phenotypes with the Tet2 knockout mice, including global 5-hmCyt reduction, altered DNA methylation, impaired hematopoietic differentiation, myeloid skewing and the development of myeloid disorders [53].The IDH2-R140Q transgenic mice showed normal HSC numbers and lineage differentiation, but exhibited increased extramedullary hematopoiesis and, when combined with HoxA9/Meis1 or FLT3 mutations in hematopoietic cells, produced acute leukemia [54].
4. Alterations within Epigenetic Regulators in MDS/AML and CLL
4.1. DNMT1
DNMT1 is significantly overexpressed in AML [55,56]. However, mutations of DNMT1 are rarely observed in contrast to the frequent appearance of DNMT3A mutations in AML and MDS [57,58] (Table 1). DNMT1 forms complexes with important transcription factors with established roles in AML, highlighting a possible pathoepigenetic interconnection in the disease biology. For example, recent studies suggest that DNMT1 transcription is modulated by the Sp1 transcription factor [59]. The regulatory functions of Sp1 are mediated through its physical interaction with other transcription factors, such as those of the NF-κB family that are constitutively activated in AML [59,60]. Such interactions were shown to lead to increased DNMT1 expression and consequently, gene silencing both in vivo and in vitro [59]. RUNX1, which is mutated in 20% of AML and MDS cases, is a weak transcription activator although, its transcriptional potential is enhanced by acting in synergy with lineage specific transcription factors. DNMT1 inhibition in RUNX1 AML restores terminal differentiation through the switch of the RUNX1-Pu.1 coactivator/corepressor activity complex via the interaction of Pu.1 with DNMT1 [61]. Minimally decreased expression of Pu.1 in mice may induce a pre-leukemic state with dysplastic features and initiate AML [62].
An important feature of AML is the existence of characteristic fusion genes, which might confer a worse (mixed lineage leukemia (MLL)-TET1/LCX fusions) or better (PML-RARα) prognosis. The PML-RARα onco-fusion induces hypermethylation by recruiting DNMT1 and DNMT3A in target promoters, such as RARb2, and is also required for the normal localization of DNMT1, which is otherwise dispersed in the nucleus [63]. Similarly, through its physical association with DNMT1, the AML-ETO fusion binds and hypermethylates AML target gene promoters [64]. DNMT1 is indirectly regulated via the miR-29b-Sp1 complex, which binds physically to the DNMT1 promoter to positively regulate its expression. Hence, there is a miR-29b-Sp1-DNMT1 axis in AML that is deregulated beginning with the underexpression of miR-29b. Forced miR-29b expression leads to restored expression of DNMT1 and its target genes p15 and ESR1, inducing partial differentiation of blast cells [65,66].
However, the role of the miR-29 family in epigenetic regulation is much more complex, because not only DNMT3A, DNMT3B, but also TET1 and thymine DNA glycosidase (TDG) are direct targets of miR-29. Furthermore, miR-29 overexpression has been shown to upregulate the global DNA methylation level in some cancer cells and downregulate DNA methylation in others [67,68].
In comparisons of DNMT1 expression in CLL with normal CD19+ B cells and memory B cells, no differences were observed [12,69]. In addition, we have previously published data demonstrating an absence of significant differences between the IGHV-UM and IGHV-M CLLs and stable expression of DNMT1 during disease progression, which further supports a minor role for DNMT1 in disease progression [12].
Table 1 Epigenetic regulators in myelodisplastic syndromes (MDS), acute myeloid leukemia (AML) and chronic lymphocytic leukemia (CLL).
|
Functions |
Genes |
MDS (mutation frequency %) |
Effect of mutations on transcription (gain/loss of function) |
AML (mutation frequency %) |
Effect of mutations on transcription (gain/loss of function) |
CLL (mutation frequency %) |
Effect of mutations on transcription (gain/loss of function) |
|
DNA methylation |
DNMT1 |
4.7 [70] |
NR |
Rare [58] |
NR |
NR |
NR |
|
DNMT3A |
7.9 [71] |
activation (loss of function) |
22.1 [72] |
activation (loss of function) |
NR |
NR |
|
|
DNA hydroxymethylation |
TET1 |
NR |
NR |
rare [73] |
NR |
2.8 [74] |
NR |
|
TET2 |
18.3 [75] |
NR (loss of function) |
12 [73] |
ND (loss of function) |
2.3 [74] |
NR |
|
|
TET3 |
NR |
NR |
rare [73] |
NR |
3.2 [74] |
NR |
|
|
IDH1 |
2.8 [70] |
NR (gain of function) |
8.9 [76] |
NR (gain of function) |
2.3 [74] |
NR |
|
|
IDH2 |
0.9 [70] |
NR (gain of function) |
10.4 [77] |
NR (gain of function) |
0.9 [74] |
NR |
DNMT, DNA methyltransferase; TET, ten-eleven translocation; IDH, isocytrate dehydrogenase; NR, not reported.
4.2. DNMT3A
DNMT3A can be mutated in both MDS and AML. DNMT3A mutations, which occur in less than 10% of MDS cases and 12%–35% of AML, are most commonly heterozygous. DNMT3A R882(C/H) represents the most frequent mutation and is associated with poorer survival [65,78,79]. DNMT3A R882H mutants show both decreased methyltransferase activity and dominant negative functions as its overexpression results in hypomethylation [80,81]. Therefore, a partial, rather than complete inactivation of DNMT3A is likely more relevant in the pathogenesis of AML/MDS. Although they occur less frequently in MDS, DNMT3A mutations may represent a risk factor for progression to AML [82]. Furthermore, several studies have provided evidence that DNMT3A mutations represent early events in leukemogenesis, and may also be present in T lymphocytes derived from AML patients. This observation shows that DNMT3A mutations are harbored within founding clones that retain the capacity to give rise to both myeloid and lymphoid lineages [83,84,85]; DNMT3A mutations are also thought to occur early in MDS [78].
However, acquired DNMT3A mutations are highly prevalent in age-associated clonal hematopoiesis in normal individuals with low or no biological impact and an, as yet, undefined oncogenic risk [86]. This suggests that mutations in epigenetic modifiers, such as DNMT3A, are permissive for leukemogenesis but not sufficient to initiate transformation alone.
The effect of DNMT3A mutations on DNA methylation in AML is controversial, possibly due to the different technical platforms used by various investigators. By using liquid chromatography tandem mass spectrometry or ELISA-based DNA methylation measurements to identify global changes in 5-mCyt, no statistical difference was detected in bulk 5-mCyt [72,87]. However, by using HM-450K BeadChip arrays and separating DNMT3A wild-type, DNMT3A R882-mutant, and DNMT3A non-R882-mutant AMLs, the R882 mutant AMLs were found to have lower mean methylation values than were detected in both the non-R882 and DNMT3A wild-type patients [81]. Hypomethylation is rather more focal than global and mainly localized in CpG islands and shores.
In CLL, several studies, including our own recently published report, have shown DNMT3A downregulation during disease progression, compared to normal CD19+ B cells or memory B cells [12,69,88]. Indeed, DNMT3A transcript levels decrease in CLL patients who progress from the indolent Binet A stage to the more aggressive Binet B/C stages [12]. On the other hand, in patients with stable disease (Binet A) for more than 5 years, there was no significant difference in the DNMT3A levels between the two time points [12]. Thus, it is unsurprising that low DNMT3A levels are associated with negative prognostic impact cytogenetic anomalies, such as 17p deletion, trisomy 12 or a complex karyotype and with a shorter treatment-free survival (TFS) [12,89]. Our group also evaluated the association of DNMT3A downregulation in the progressive CLL disease group with changes in 5-mCyt levels by using the base resolution sequencing technique, RRBS, which revealed a significant decrease in DNA 5-mCyt in all regions of the genome (unpublished data). These findings support the view that loss in DNMT3A activity represents a driver for MDS/AML progression, and a risk factor for disease activity in CLL as reported previously in the DNMT3a+/- and Eμ-DNMT3a-/- mice, respectively.
4.3. TET1
TET1 was first identified as a fusion partner, TET1/LCX, of the MLL gene in AML carrying t(10,11)(q22;q23). Furthermore, TET1 represents a direct target of MLL-fusion proteins, such as HOXA9, MEIS1, and PBX3, and is significantly upregulated in MLL-rearranged leukemia, leading to a global increase in 5-hmCyt levels [90]. In normal cytogenetic AML, TET1 overexpression is associated with poor survival [91].
In CLL, somatic mutations in the TET1 gene represent rare events (2.8%) and CLL B cells have similar transcriptional levels of TET1 to those observed in control memory B cells [12,74]. However, low levels of TET1 are associated with shorter TFS [12].
4.4. TET2
TET2 is mutated in approximately 12%–20% of AMLs, 23%–35% of MDS and 50% of CMML [92,93]. These loss-of-function mutations are diverse and impair TET2 activity [93,94]. In primary MDS and AML patients harboring TET2 mutations, significantly reduced global 5-hmCyt levels have been revealed [87,95].
However, TET2 mutations, like those present in DNMT3A, are frequent in age-associated clonal hematopoiesis in normal individuals with low or no biological impact, which shows these mutations are not sufficient to initiate leukemogenesis alone [86].
In CLL, only 3.2% of patients present somatic mutations in TET2. In the early stage of the disease TET2 transcript levels in CLL are similar to those of control memory B cells, while important variations in TET2 levels are reported between CLL patients [12,73]. In these two reports, a reduction in TET2 is associated with a lower median TFS, supporting an unexpected role for TET2 as an oncogenic repressor.
4.5. Isocitrate Dehydrogenase-1 (IDH1) and -2 (IDH2)
The isocitrate dehydrogenase-1 (IDH1) and -2 (IDH2) genes encode NADP+ dependent enzymes that convert isocitrate to α-KT in the Krebs cycle. IDH1 functions in the cytoplasm whereas IDH2 is localized to the mitochondria. Mutations in the genes encoding IDH1 and IDH2 tend not to occur together in the same clone and have been described in 15%–30% of AMLs [96]. Mutations result in a new enzymatic function resulting in the aberrantly accelerated conversion of α-KTG to 2-HG [97]. The accumulation of the metabolite 2-HG has been shown to be responsible for the inhibition of TET2 function, thus ultimately inhibiting hydroxymethylation of DNA. Mutations in IDH1/2 and TET2 mutations are mutually exclusive in AML and, as expected, patients show similar DNA methylation profiles [98]. While the prognostic impact of IDH mutations in AML is controversial and remains unclear, clinical trials using inhibitors that are relatively specific for IDH1 and IDH2 are ongoing [99,100,101].
In CLL, IDH1/2 mutations are extremely rare (0.9%) and decreased expression of IDH2 in addition to IDH1 overexpression were revealed in CLL B cells compared to the levels detected in normal CD19+ B cells [74].
5. Conclusions
Advances in gene expression profiling and the development of new and improved high-throughput techniques for exploring methylation and hydroxymethylation of DNA have provided a more accurate and detailed understanding of the genetic and epigenetic basis for the development of hematopoietic diseases. Since their discovery, many important advances have also been made in our mechanistic understanding of the enzymes implicated in DNA methylation and demethylation processes.
However, although these enzymatic processes have been thoroughly characterized and high-resolution co-crystal structures of human DNMTs and TETs and their substrates have provided structural insights into their activities, several fundamental aspects of these processes and their implications in hematologic myeloid and lymphoid malignancies remain to be fully elucidated. First, concurrent recruitment of auxiliary factors by DNMT and TET proteins to genomic target sites is far from clear at the moment. Although several interactions have been demonstrated, studying these interactions in association with specific oncogenes or tumor suppressor genes implicated in hematologic malignancies could provide important information concerning their roles in these pathologies.
Second, the mechanisms that govern recruitment and localization of DNMTs and TETs to DNA are not fully understood. Addressing these questions will be an important part of future investigations.
We chose to discuss epigenetic modifications from two perspectives – the myeloid lineage pathologies, MDS/AML, in which epigenetic regulators are dysregulated because they present somatic mutations, and the lymphoid lineage pathology, CLL, in which epigenetic regulators are rarely mutated, but are highly dysregulated (Figure 3). With regard to MDS/AML, epigenetic dysregulation is an early event in the development of HSC. In CLL, on the other hand, there is a step-wise process of leukemogenesis and disease progression, and epigenetic regulators and methylation have an important dynamic correlation with disease progression and prognosis.
Irrespective of the underlying molecular events, DNMT3A and TET2 in particular are dysregulated in MDS/AML and CLL, leading to altered DNA methylation and hydroxymethylation pattern. Advances in gene expression profiling and the development of new and improved high-throughput techniques for exploring methylation and hydroxymethylation of DNA have provided a more accurate and detailed understanding of the genetic and epigenetic basis for the development of hematopoietic diseases. Since their discovery, many important advances have also been made in our mechanistic understanding of the enzymes implicated in DNA methylation and demethylation processes.
However, although these enzymatic processes have been thoroughly characterized and high-resolution co-crystal structures of human DNMTs and TETs and their substrates have provided structural insights into their activities, several fundamental aspects of these processes and their implications in hematologic myeloid and lymphoid malignancies remain to be fully elucidated. First, concurrent recruitment of auxiliary factors by DNMT and TET proteins to genomic target sites is far from clear at the moment. Although several interactions have been demonstrated, studying these interactions in association with specific oncogenes or tumor suppressor genes implicated in hematologic malignancies could provide important information concerning their roles in these pathologies.
Second, the mechanisms that govern recruitment and localization of DNMTs and TETs to DNA are not fully understood. Addressing these questions will be an important part of future investigations.
We chose to discuss epigenetic modifications from two perspectives – the myeloid lineage pathologies, MDS/AML, in which epigenetic regulators are dysregulated because they present somatic mutations, and the lymphoid lineage pathology, CLL, in which epigenetic regulators are rarely mutated, but are highly dysregulated (Figure 3). With regard to MDS/AML, epigenetic dysregulation is an early event in the development of HSC. In CLL, on the other hand, there is a step-wise process of leukemogenesis and disease progression, and epigenetic regulators and methylation have an important dynamic correlation with disease progression and prognosis.
Irrespective of the underlying molecular events, DNMT3A and TET2 in particular are dysregulated in MDS/AML and CLL, leading to altered DNA methylation and hydroxymethylation patterns that confer a competitive advantage to the myeloid or lymphoid lineage and an imbalance between self-renewal and differentiation. A better understanding of their localization and their co-recruitment of other proteins at specific DNA target sites could provide us with the possibility to modulate hematopoiesis, restore the initial balance, and control disease progressions that confer a competitive advantage to the myeloid or lymphoid lineage and an imbalance between self-renewal and differentiation. A better understanding of their localization and their co-recruitment of other proteins at specific DNA target sites could provide us with the possibility to modulate hematopoiesis, restore the initial balance, and control disease progression.
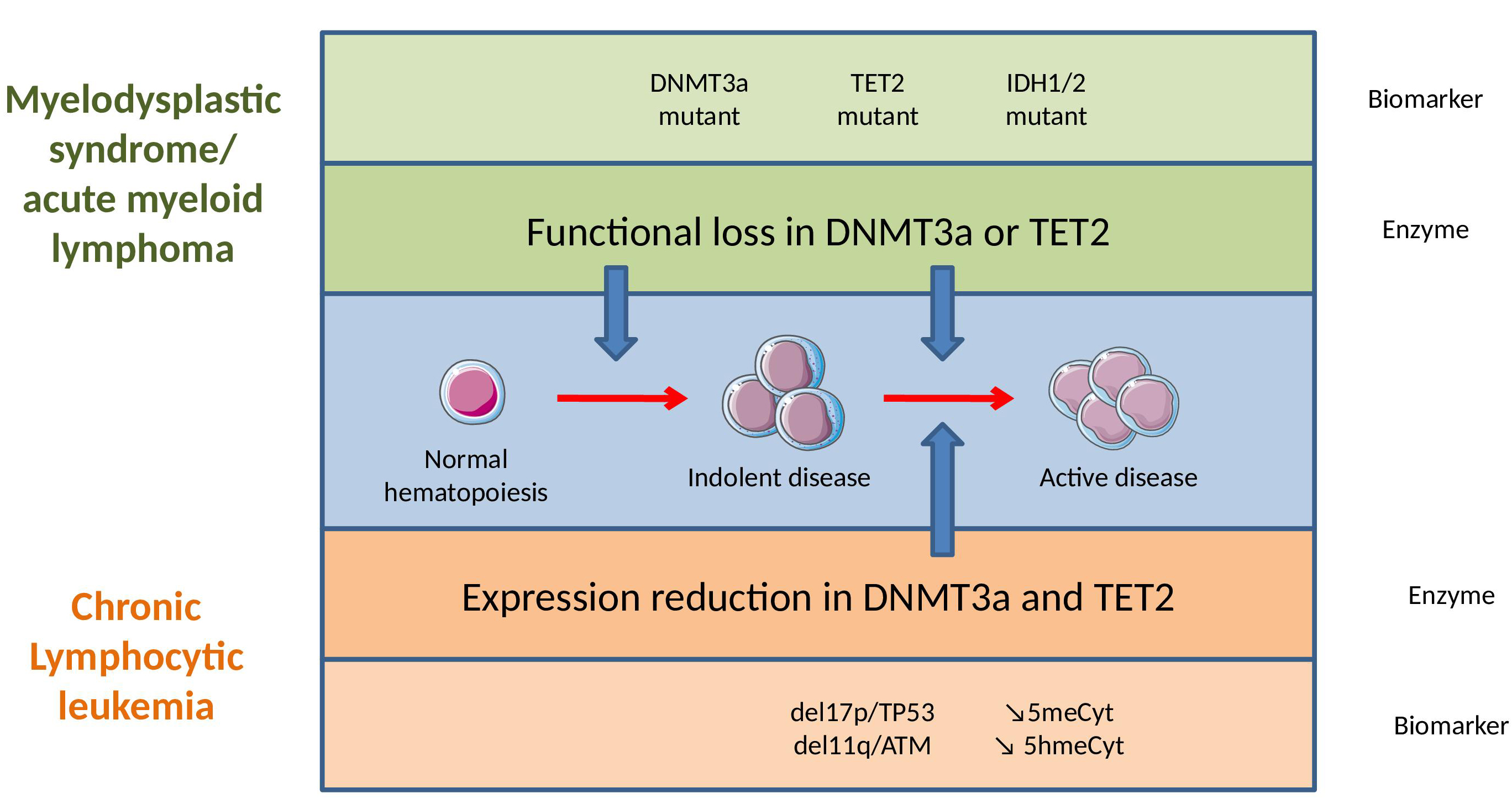
Figure 3 Epigenetic dysregulation in myelodysplastic syndrome (MDS)/acute myeloid leukemia (AML) and in chronic lymphocytic leukemia (CLL).
(Note: In MDS and AML, epigenetic dysregulation is an early event caused by somatic mutations in hematopoietic stem cells, which generate a loss of function at the protein level and a poor prognosis. In CLL, epigenetic regulators are rarely mutated, but highly dysregulated, with their expression diminishing in association with disease progression.)
Acknowledgments
Authors express their gratitude to Simone Forest and Geneviève Michel for their help in editing the paper. This study was supported by research funding from the “Association Laurette Fugain” (ALF 2015/03), the “ligue against the cancer sections 29/35/49, the "Region Bretagne" and from the REpiCGO an Epigenetic network of the "Cancéropole Grand Ouest".
Author Contributions
The original manuscript was written by CBa and YR (main expertise in epigenetics/CLL) and the other authors (main expertise in MDS/AML and/or epigenetics) modified and approved the final manuscript.
Competing Interests
The authors have declared that no competing interests exist.
References
- Shallis RM, Ahmad R, Zeidan AM. The genetic and molecular pathogenesis of myelodysplastic syndromes. Eur J Haematol. 2018; 101: 260-271. [CrossRef] [Google scholar] [PubMed]
- Gilliland DG, Tallman MS. Focus on acute leukemias. Cancer Cell. 2002; 1: 417-420. [CrossRef] [Google scholar] [PubMed]
- Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood. 2012;119: 34-43. [CrossRef] [Google scholar] [PubMed]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. “Cancer statistics, 2007. CA Cancer J Clin. 2007; 57: 43-66. [CrossRef] [Google scholar] [PubMed]
- Hallek M. Chronic lymphocytic leukemia: 2017 update on diagnosis, risk stratification, and treatment. Am J Hematol. 2017; 92: 946-965. [CrossRef] [Google scholar] [PubMed]
- Renadineau Y, Bariller E, Pers JO. B1‐ and CD5‐ Positive B Cells. eLS. 2014; DOI: 10.1002/9780470015902.a0024242. [CrossRef] [Google scholar]
- Klein U, Tu Y, Stolovitzky GA, Mattioli M, Cattoretti G, Husson H, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001; 194: 1625-1638. [CrossRef] [Google scholar] [PubMed]
- Oakes CC, Seifert M, Assenov Y, Gu L, Przekopowitz M, Ruppert AS, et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet. 2016; 48: 253-264. [CrossRef] [Google scholar] [PubMed]
- Kikushige Y, Ishikawa F, Miyamoto T, Shima T, Urata S, Yoshimoto G, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011; 20: 246-259. [CrossRef] [Google scholar] [PubMed]
- Garaud S, Le Dantec C, Berthou C, Lydyard PM, Youinou P, Renaudineau Y. Selection of the alternative exon 1 from the cd5 gene down-regulates membrane level of the protein in B lymphocytes. J Immunol. 2008; 181: 2010-2018. [CrossRef] [Google scholar] [PubMed]
- Mankai A, Bordron A, Renaudineau Y, Martins-Carvalho C, Takahashi S, Ghedira I, et al. Purine-rich box-1-mediated reduced expression of CD20 alters rituximab-induced lysis of chronic lymphocytic leukemia B cells. Cancer Res. 2008; 68: 7512-7519. [CrossRef] [Google scholar] [PubMed]
- Bagacean C, Tempescul A, Le Dantec C, Bordron A, Mohr A, Saad H, et al. Alterations in DNA methylation/demethylation intermediates predict clinical outcome in chronic lymphocytic leukemia. Oncotarget. 2017; 8: 65699-65716. [CrossRef] [Google scholar] [PubMed]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009; 324: 929-930. [CrossRef] [Google scholar] [PubMed]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009; 324: 930-935. [CrossRef] [Google scholar] [PubMed]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009; 462: 315-322. [CrossRef] [Google scholar] [PubMed]
- Renaudineau Y, Hillion S, Saraux A, Mageed RA, Youinou P. An alternative exon 1 of the CD5 gene regulates CD5 expression in human B lymphocytes. Blood. 2005; 106: 2781-2789. [CrossRef] [Google scholar] [PubMed]
- Renaudineau Y, Vallet S, Le Dantec C, Hillion S, Saraux A, Youinou P. Characterization of the human CD5 endogenous retrovirus-E in B lymphocytes. Genes Immun. 2005; 6: 663-671. [CrossRef] [Google scholar] [PubMed]
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013; 14: 204-220. [CrossRef] [Google scholar] [PubMed]
- Le Dantec C, Vallet S, Brooks WH, Renaudineau Y. Human endogenous retrovirus group E and its involvement in diseases. Viruses. 2015; 7: 1238-1257. [CrossRef] [Google scholar] [PubMed]
- Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND, et al. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One. 2011; 6: e14524. [CrossRef] [Google scholar] [PubMed]
- Brooks WH, Renaudineau Y. Epigenetics and autoimmune diseases: the X chromosome-nucleolus nexus. Front Genet. 2015; 6: 22. [CrossRef] [Google scholar] [PubMed]
- Charras A, Konsta OD, Le Dantec C, Bagacean C, Kapsogeorgou EK, Tzioufas AG, et al. Cell-specific epigenome-wide DNA methylation profile in long-term cultured minor salivary gland epithelial cells from patients with Sjogren's syndrome. Ann Rheum Dis. 2017; 76: 625-628. [CrossRef] [Google scholar] [PubMed]
- Madakashira BP, Sadler KC. DNA Methylation, Nuclear Organization, and Cancer. Front Genet. 2017; 8: 76. [CrossRef] [Google scholar] [PubMed]
- Brandeis M, Frank D, Keshet I, Siegfried Z, Mendelsohn M, Nemes A, et al. Sp1 elements protect a CpG island from de novo methylation. Nature. 1994; 371: 435-438. [CrossRef] [Google scholar] [PubMed]
- Macleod D, Charlton J, Mullins J, Bird AP. Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev. 1994; 8: 2282-2292. [CrossRef] [Google scholar] [PubMed]
- Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. 2001; 20: 3139-3155. [CrossRef] [Google scholar] [PubMed]
- Brooks WH, Le Dantec C, Pers JO, Youinou P, Renaudineau Y. Epigenetics and autoimmunity. J Autoimmun. 2010; 34: J207-219. [CrossRef] [Google scholar] [PubMed]
- Bestor TH, Ingram VM. Two DNA methyltransferases from murine erythroleukemia cells: purification, sequence specificity, and mode of interaction with DNA. Proc Natl Acad Sci USA. 1983; 80: 5559-5563. [CrossRef] [Google scholar] [PubMed]
- Bestor TH. Cloning of a mammalian DNA methyltransferase. Gene. 1988; 74: 9-12. [CrossRef] [Google scholar] [PubMed]
- Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998; 19: 219-220. [CrossRef] [Google scholar] [PubMed]
- Deplus R, Brenner C, Burgers WA, Putmans P, Kouzarides T, de Launoit Y, et al. Dnmt3L is a transcriptional repressor that recruits histone deacetylase. Nucleic Acids Res. 2002; 30: 3831-3838. [CrossRef] [Google scholar] [PubMed]
- Wyatt GR, Cohen SS. A new pyrimidine base from bacteriophage nucleic acids. Nature. 1952; 170: 1072-1073. [CrossRef] [Google scholar] [PubMed]
- Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016; 30: 733-750. [CrossRef] [Google scholar] [PubMed]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011; 333: 1300-1303. [CrossRef] [Google scholar] [PubMed]
- Bagacean C, Zdrenghea M, Dantec CL, Tempescul A, Berthou C, Renaudineau Y. DNA demethylation marks in chronic lymphocytic leukemia: it is time to let the cat out of the bag. Future Sci OA. 2018; 4: FSO265. [Google scholar]
- Nestor CE, Ottaviano R, Reddington J, Sproul D, Reinhardt D, Dunican D, et al. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012; 22: 467-477. [CrossRef] [Google scholar] [PubMed]
- Zhang X, Su J, Jeong M, Ko M, Huang Y, Park HJ, et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat Genet. 2016; 48: 1014-1023. [CrossRef] [Google scholar] [PubMed]
- Huang Y, Chavez L, Chang X, Wang X, Pastor WA, Kang J, et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc Natl Acad Sci USA. 2014; 111: 1361-1366. [CrossRef] [Google scholar] [PubMed]
- Gu T, Lin X, Cullen SM, Luo M, Jeong M, Estecio M, et al. DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biol. 2018; 19: 88. [CrossRef] [Google scholar] [PubMed]
- Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013; 32: 645-655. [CrossRef] [Google scholar] [PubMed]
- Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011; 473: 343-348. [CrossRef] [Google scholar] [PubMed]
- Bock C, Beerman I, Lien WH, Smith ZD, Gu H, Boyle P, et al. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell. 2012; 47: 633-647. [CrossRef] [Google scholar] [PubMed]
- Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010; 467: 338-342. [CrossRef] [Google scholar] [PubMed]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011; 44: 23-31. [CrossRef] [Google scholar] [PubMed]
- Challen GA, Sun D, Mayle A, Jeong M, Luo M, Rodriguez B, et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell. 2014; 15: 350-364. [CrossRef] [Google scholar] [PubMed]
- Cole CB, Russler-Germain DA, Ketkar S, Verdoni AM, Smith AM, Bangert CV, et al. Haploinsufficiency for DNA methyltransferase 3A predisposes hematopoietic cells to myeloid malignancies. J Clin Invest. 2017; 127: 3657-3674. [CrossRef] [Google scholar] [PubMed]
- Peters SL, Hlady RA, Opavska J, Klinkebiel D, Pirruccello SJ, Talmon GA, et al. Tumor suppressor functions of Dnmt3a and Dnmt3b in the prevention of malignant mouse lymphopoiesis. Leukemia. 2014; 28: 1138-1142. [CrossRef] [Google scholar] [PubMed]
- Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y, et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol. 2015; 16: 653-662. [CrossRef] [Google scholar] [PubMed]
- Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011; 118: 4509-4518. [CrossRef] [Google scholar] [PubMed]
- Mouly E, Ghamlouch H, Della-Valle V, Scourzic L, Quivoron C, Roos-Weil D, et al. B-cell tumor development in Tet2-deficient mice. Blood Adv. 2018; 2: 703-714. [CrossRef] [Google scholar] [PubMed]
- An J, Gonzalez-Avalos E, Chawla A, Jeong M, Lopez-Moyado IF, Li W, et al. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat Commun. 2015; 6: 10071. [CrossRef] [Google scholar] [PubMed]
- Zhao Z, Chen L, Dawlaty MM, Pan F, Weeks O, Zhou Y, et al. Combined Loss of Tet1 and Tet2 Promotes B Cell, but Not Myeloid Malignancies, in Mice. Cell Rep. 2015; 13: 1692-1704. [CrossRef] [Google scholar] [PubMed]
- Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013; 3: 730-741. [CrossRef] [Google scholar] [PubMed]
- Kats LM, Reschke M, Taulli R, Pozdnyakova O, Burgess K, Bhargava P, et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell. 2014; 14: 329-341. [CrossRef] [Google scholar] [PubMed]
- Mizuno S, Chijiwa T, Okamura T, Akashi K, Fukumaki Y, Niho Y, et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood. 2001; 97: 1172-1179. [CrossRef] [Google scholar] [PubMed]
- Langer F, Dingemann J, Kreipe H, Lehmann U. Up-regulation of DNA methyltransferases DNMT1, 3A, and 3B in myelodysplastic syndrome. Leuk Res. 2005; 29: 325-329. [CrossRef] [Google scholar] [PubMed]
- Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, Mauch J, Kelkenberg-Schade S, Haldemann B, et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood. 2012; 120: e83-92. [CrossRef] [Google scholar] [PubMed]
- Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013; 368: 2059-2074. [CrossRef] [Google scholar] [PubMed]
- Liu S, Liu Z, Xie Z, Pang J, Yu J, Lehmann E, et al. Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/ NF-kappaB-dependent DNA methyltransferase activity in acute myeloid leukemia. Blood. 2008; 111: 2364-2373. [CrossRef] [Google scholar] [PubMed]
- Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J. 1993; 12: 3551-3558. [CrossRef] [Google scholar] [PubMed]
- 61. Gu X, Hu Z, Ebrahem Q, Crabb JS, Mahfouz RZ, Radivoyevitch T, et al. Runx1 regulation of Pu.1 corepressor/coactivator exchange identifies specific molecular targets for leukemia differentiation therapy. J Biol Chem. 2014; 289: 14881-14895. [CrossRef] [Google scholar] [PubMed]
- Will B, Vogler TO, Narayanagari S, Bartholdy B, Todorova TI, da Silva Ferreira M, et al. Minimal PU.1 reduction induces a preleukemic state and promotes development of acute myeloid leukemia. Nat Med. 2015; 21: 1172-1181. [CrossRef] [Google scholar] [PubMed]
- Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science. 2002; 295: 1079-1082. [CrossRef] [Google scholar] [PubMed]
- Liu S, Shen T, Huynh L, Klisovic MI, Rush LJ, Ford JL, et al. Interplay of RUNX1/MTG8 and DNA methyltransferase 1 in acute myeloid leukemia. Cancer Res. 2005; 65: 1277-1284. [CrossRef] [Google scholar] [PubMed]
- Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009; 113: 6411-6418. [CrossRef] [Google scholar] [PubMed]
- Bernot KM, Nemer JS, Santhanam R, Liu S, Zorko NA, Whitman SP, et al. Eradicating acute myeloid leukemia in a Mll(PTD/wt): Flt3(ITD/wt) murine model: a path to novel therapeutic approaches for human disease. Blood.2013; 122: 3778-3783. [CrossRef] [Google scholar] [PubMed]
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B.“ Proc Natl Acad Sci USA. 2007; 104: 15805-15810. [CrossRef] [Google scholar] [PubMed]
- Morita S, Horii T, Kimura M, Ochiya T, Tajima S, Hatada I. MiR-29 represses the activities of DNA methyltransferases and DNA demethylases. Int J Mol Sci. 2013; 14: 14647-14658. [CrossRef] [Google scholar] [PubMed]
- Kn H, Bassal S, Tikellis C, El-Osta A. Expression analysis of the epigenetic methyltransferases and methyl-CpG binding protein families in the normal B-cell and B-cell chronic lymphocytic leukemia (CLL). Cancer Biol Ther. 2004; 3: 989-994. [CrossRef] [Google scholar] [PubMed]
- Jung SH, Kim YJ, Yim SH, Kim HJ, Kwon YR, Hur EH, et al. Somatic mutations predict outcomes of hypomethylating therapy in patients with myelodysplastic syndrome. Oncotarget. 2016; 7: 55264-55275. [CrossRef] [Google scholar] [PubMed]
- Lin ME, Hou HA, Tsai CH, Wu SJ, Kuo YY, Tseng MH, et al. Dynamics of DNMT3A mutation and prognostic relevance in patients with primary myelodysplastic syndrome. Clin Epigenetics. 2018; 10: 42. [CrossRef] [Google scholar] [PubMed]
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010; 363: 2424-2433. [CrossRef] [Google scholar] [PubMed]
- Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009; 114: 144-147. [CrossRef] [Google scholar] [PubMed]
- Van Damme M, Crompot E, Meuleman N, Maerevoet M, Mineur P, Bron D, et al. Characterization of TET and IDH gene expression in chronic lymphocytic leukemia: comparison with normal B cells and prognostic significance. Clin Epigenetics. 2016; 8: 132. [CrossRef] [Google scholar] [PubMed]
- Guo Z, Zhang SK, Zou Z, Fan RH, Lyu XD. Prognostic significance of TET2 mutations in myelodysplastic syndromes: A meta-analysis. Leuk Res. 2017; 58: 102-107. [CrossRef] [Google scholar] [PubMed]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009; 361: 1058-1066. [CrossRef] [Google scholar] [PubMed]
- Chotirat S, Thongnoppakhun W, Promsuwicha O, Boonthimat C, Auewarakul CU. Molecular alterations of isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) metabolic genes and additional genetic mutations in newly diagnosed acute myeloid leukemia patients. J Hematol Oncol. 2012; 5: 5. [CrossRef] [Google scholar] [PubMed]
- Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011; 25: 1153-1158. [CrossRef] [Google scholar] [PubMed]
- Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011; 43: 309-315. [CrossRef] [Google scholar] [PubMed]
- Kim SJ, Zhao H, Hardikar S, Singh AK, Goodell MA, Chen T. A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood. 2013; 122: 4086-4089. [CrossRef] [Google scholar] [PubMed]
- Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell. 2014; 25: 442-454. [CrossRef] [Google scholar] [PubMed]
- Thol F, Winschel C, Ludeking A, Yun H, Friesen I, Damm F, et al. Rare occurrence of DNMT3A mutations in myelodysplastic syndromes. Haematologica. 2011; 96: 1870-1873. [CrossRef] [Google scholar] [PubMed]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012; 481: 506-510. [CrossRef] [Google scholar] [PubMed]
- Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA. 2014; 111: 2548-2553. [CrossRef] [Google scholar] [PubMed]
- Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014; 506: 328-333. [CrossRef] [Google scholar] [PubMed]
- Buscarlet M, Provost S, Zada YF, Barhdadi A, Bourgoin V, Lepine G, et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. 2017; 130: 753-762. [CrossRef] [Google scholar] [PubMed]
- Kroeze LI, Aslanyan MG, van Rooij A, Koorenhof-Scheele TN, Massop M, Carell T, et al. Characterization of acute myeloid leukemia based on levels of global hydroxymethylation. Blood. 2014; 124: 1110-1118. [CrossRef] [Google scholar] [PubMed]
- Wang J, Morrissette J, Lieberman DB, Timlin C, Schuster SJ, Mato AR. Utilization of next generation sequencing identifies potentially actionable mutations in chronic lymphocytic leukaemia. Br J Haematol. 2018; 180: 299-301. [CrossRef] [Google scholar] [PubMed]
- Bagacean C, Le Dantec C, Berthou C, Tempescul A, Saad H, Bordron A, et al. Combining cytogenetic and epigenetic approaches in chronic lymphocytic leukemia improves prognosis prediction for patients with isolated 13q deletion. Clin Epigenetics. 2017; 9: DOI: 10.1186/s13148-017-0422-7. [CrossRef] [Google scholar] [PubMed]
- Huang H, Jiang X, Li Z, Li Y, Song CX, He C, et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci USA. 2013; 110: 11994-11999. [CrossRef] [Google scholar] [PubMed]
- Wang J, Li F, Ma Z, Yu M, Guo Q, Huang J, et al. High Expression of TET1 Predicts Poor Survival in Cytogenetically Normal Acute Myeloid Leukemia From Two Cohorts. EBioMedicine. 2018; 28: 90-96. [CrossRef] [Google scholar] [PubMed]
- Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012; 366: 1079-1089. [CrossRef] [Google scholar] [PubMed]
- Meldi KM, Figueroa ME. Cytosine modifications in myeloid malignancies. Pharmacol Ther. 2015; 152: 42-53. [CrossRef] [Google scholar] [PubMed]
- Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009; 360: 2289-2301. [CrossRef] [Google scholar] [PubMed]
- Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014; 9: 1841-1855. [CrossRef] [Google scholar] [PubMed]
- Ohgami RS, Arber DA. The diagnostic and clinical impact of genetics and epigenetics in acute myeloid leukemia. Int J Lab Hematol. 2015; 37: 122-132. [CrossRef] [Google scholar] [PubMed]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010; 17: 225-234. [CrossRef] [Google scholar] [PubMed]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010; 18: 553-567. [CrossRef] [Google scholar] [PubMed]
- Boissel N, Nibourel O, Renneville A, Gardin C, Reman O, Contentin N, et al. Prognostic impact of isocitrate dehydrogenase enzyme isoforms 1 and 2 mutations in acute myeloid leukemia: a study by the Acute Leukemia French Association group. J Clin Oncol. 2010; 28: 3717-3723. [CrossRef] [Google scholar] [PubMed]
- Koszarska M, Bors A, Feczko A, Meggyesi N, Batai A, Csomor J, et al. Type and location of isocitrate dehydrogenase mutations influence clinical characteristics and disease outcome of acute myeloid leukemia. Leuk Lymphoma. 2013; 54: 1028-1035. [CrossRef] [Google scholar] [PubMed]
- Gallipoli P, Giotopoulos G, Huntly BJ. Epigenetic regulators as promising therapeutic targets in acute myeloid leukemia. Ther Adv Hematol. 2015; 6: 103-119. [CrossRef] [Google scholar] [PubMed]

