

Carrier Screening for the Haemoglobinopathies: Past, Present and Future
John Old 1,* ![]() , Cornelis Harteveld 2
, Cornelis Harteveld 2![]()
- National Haemoglobinopathy Reference Laboratory, Biomedical Research Centre Molecular Diagnostic, Laboratory, Haematology Department, John Radcliffe Hospital, Oxford, UK
- Department of Clinical Genetics, Haemoglobinopathies Expert Centre, Leiden University Medical Centre, Einthovenweg 20, 2333ZC Leiden, The Netherlands
* Correspondence: John Old ![]() .
.
Received: July 03, 2017 | Accepted: August 07, 2017 | Published: August 15, 2017
OBM Genetics 2017, Volume 1, Issue 3, doi:10.21926/obm.genet.1703005
Academic Editors: Joanne Traeger-Synodinos and François Rousseau
Recommended citation: Old J, Harteveld C. Carrier Screening for the Haemoglobinopathies: Past, Present and Future. OBM Genetics 2017;1(3):005; doi:10.21926/obm.genet.1703005.
© 2017 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Carrier screening for the haemoglobinopathies has undergone many technological improvements in haematological and molecular diagnostic techniques since the first prenatal diagnoses by DNA analysis in the 1970s by Southern blot analysis enabled the implementation of effective successful prevention programmes for beta thalassaemia involving public education, carrier screening, genetic counselling and prenatal diagnosis in Mediterranean countries. The application of a wide variety of PCR-based molecular diagnostic techniques which allows the detection of the complete range of haemoglobinopathy mutations has led to the establishment of comprehensive national prevention programmes in many developing countries and also in countries such as those in Northern Europe in which the prevalence and heterogeneity of the haemoglobinopathies has been significantly increased by population immigration. Despite the great technological advances in mutation detection, the screening of haemoglobinopathies still requires the combined use of haematological and molecular techniques to arrive at an accurate diagnosis, and requires specialist knowledge of genotype/phenotype relationships because of the multitude of complex phenotypes which result from interactions between genotypes and co-inherited globin gene disorders relationships. The latest technological advances in mutation analysis techniques and the application of some of these for the noninvasive approach of analysis of fetal DNA in maternal blood are anticipated to improve haemoglobinopathy prevention programmes in the future.
Keywords
Haemoglobinopathies; thalassaemias; carrier screening; diagnosis
Introduction
The haemoglobinopathies and globin gene disorders constitute the commonest recessive monogenic disorders worldwide [1]. They are caused by more than 1700 different mutations which either affect the synthesis of globin chains (the thalassaemias) or alter the structure and properties of haemoglobin (haemoglobin variants or abnormal haemoglobins) [2]. They are mostly autosomal recessive disorders and heterozygotes are symptom-free but present various haematological characteristics which are used for their identification in carrier screening programmes. The homozygous states and compound-heterozygous states result in four main groups of clinically significant conditions, each with a variable degree of phenotypic severity: the β-thalassaemias, α-thalassaemias, sickle cell syndromes, and Hb E syndromes [3].
Many patients with these conditions are born in developing and low-income countries where they create an enormous health burden [4]. Therefore prevention of the homozygous states for β-thalassaemia and Hb S constitutes a major component of the management of these disorders in many countries, involving carrier detection by haematology screening, molecular diagnostics, genetic counseling, and prenatal diagnosis or preimplantation genetic diagnosis where applicable [5]. In the future, the recent studies involving fetal DNA in maternal plasma and the application of the latest sensitive technologies such as digital PCR and next generation sequencing may allow the routine prenatal diagnosis of globin gene disorders that is noninvasive for the fetus.
The haemoglobinopathies are a heterogeneous group of disorders, because of the numerous types of thalassaemia and abnormal haemoglobin genotypes which can interact when co-inherited, creating a complex range of haematological phenotypes that are often difficult to interpret accurately. Moreover, some phenotypes can arise from several different genotypes, such as heterozygous alpha zero thalassaemia and homozygous alpha plus thalassaemia, and the genotypes cannot be distinguished by simple haematological parameters. Finally, the electrophoresis or chromatography techniques used to screen for abnormal haemoglobins only provide a presumed diagnosis for the variant, and further test are required if a definitive diagnosis is required. Thus in many cases of carrier screening, an accurate diagnosis requires expertise in the interpretation of the haematological results and confirmation of the genotypes by DNA analysis or mass spectrometry [6]. This review of carrier screening for the haemoglobinopathies includes a discussion of the past techniques, the current techniques that are in widespread use, and finally the latest developments that will inform the approaches to haematological and molecular carrier screening and prenatal diagnosis in the future.
Screening Programmes
Successful nationwide programmes for the carrier screening of β-thalassaemia on a voluntary basis have been established since the 1970s in several Mediterranean countries with a high prevalence of homozygous beta thalassaemia, in particular in Cyprus [7] Sardinia [8] and Greece [9]. Italy, Greece, and Cyprus aimed for premarital and preconception screening, ie prospective screening and not antenatal. However, because of migration, the carrier rate of β-thalassaemia is increasing in countries that previously had a low prevalence of haemoglobinopathies, and national voluntary programmes of carrier screening and prevention for thalassaemia and sickle cell disease have now been successfully established in several European non-endemic countries such as the UK [10]. Although guidelines and recommendations regarding the conduct of carrier screening programmes have been developed by organisations such as the World Health Organisation [11,12], reviews of those conducted around the world have shown that no one strategy has met the needs of every country [13,14]. However whatever the strategy, community awareness and public education play a very important part of all prospective carrier screening programmes.
For most of these established programmes, the screening is prospective, ie when carriers are identified before having an affected child. However screening may also be retrospective – that is, when couples already have an affected child, as in the case of the Sardinia programme, in which cascade screening (or extended family testing) was used to test relatives of identified carriers and/or patients. This policy has led to the detection of 90% of expected at-risk couples through tests on only 15% of the adult population [14].
Numerous prospective carrier screening programmes are now conducted around the world. They can be divided into mandatory or voluntary programmes. Despite the WHO recommendation that no compulsory genetic testing should be carried out, some countries, including Iran, Saudi Arabia and Palestinian territories have laws in place making haemoglobinopathy screening mandatory for all couples before having the approval to get married. In Cyprus, couples waiting to get married are required by the church to be screened, counselled and have the laboratory result report. The same mandatory rule is applied in Islamic countries like Iran and the UAE’s before legal marriage. While in Cyprus there is no interference on partner choice, in Iran adapting partner choice has been advised as a first option and PD with legal medical abortion as a second. In the UAE’s adapting partner choice is the only advice provided. In other countries, including Sardinia, Greece, and in England, haemoglobinopathy screening is offered on a voluntary basis.
Antenatal Screening
Community control of sickle cell anaemia and thalassaemia by antenatal screening and fetal diagnosis requires establishment of programmes for public education and awareness, carrier screening to identify couples at risk of having a child affected with a clinically significant haemoglobinopathy, and genetic counselling in order to offer prenatal diagnosis or pre-implantation genetic diagnosis. Comprehensive programmes have been established in many countries, including Italy, Greece, Cyprus, UK, France, Turkey, Tunisia, Iran, Thailand, Australia, Singapore, Taiwan, Hong Kong, and Cuba. In addition, partial programmes including antenatal screening according to ethnic origin are available in several countries in Northern Europe (Netherlands, Belgium, and Germany) and in many developing countries such as Iran, India, and Pakistan, where problems of low resources and religious dilemmas have had to be solved, a prenatal diagnosis service has also been introduced [5].
There are two approaches to the delivery of a screening programme designed to detect carriers of β-thalassaemia, α-thalassaemia and the clinically significant haemoglobin variants: 1) a primary screen to determine red cell indices, followed by a secondary screen involving haemoglobin analysis in subjects with reduced mean corpuscular volume (MCV) and/or mean corpuscular haemoglobin (MCH), and subjects identified to be at high risk of carry such a clinically significant variant by their ethnic origin; 2) a complete screening based on determining red cell indices, haemoglobin pattern analysis and Hb A2 in all subjects from the outset. Once the wife is found to be a carrier, her partner is then subjected to the same screening approach.
A primary screening approach works best in countries with low frequency and limited heterogeneity of thalassaemia, while complete screening is best in populations where both α- and β-thalassaemias are common, and where interaction of α- and β-thalassaemias could lead to missed diagnoses due to the normalisation of red cell indices. The UK antenatal screening programme combines both approaches, with a primary screening approach used for areas with a low prevalence of Hb S and a complete screening approach for high prevalence areas (defined as an area where the estimated fetal prevalence of sickle cell disease is 1.5 per 10,000 pregnancies or greater), as detailed in the Programme’s laboratory handbook [15].
Neonatal Screening
Neonatal screening is relevant to areas where sickle cell disease is prevalent and where the at-risk couples are not detected by a population-based carrier screening programme.The early detection of affected children with sickle cell anaemia enables timely interventions which reduce the likelihood of life threatening complications such as pneumococcal infections. In addition, neonatal screening provides invaluable epidemiological information regarding the frequency and geographical distribution of homozygous sickle cell disease, sickle cell thalassaemia, Hb S/D, Hb S/C disease and Hb S/O-Arab. Despite its usefulness, practical application is limited to few countries which include the USA [16], Jamaica [17], Brazil [18], India [19] and some European countries [20]. In many high prevalence areas, including Africa and South America, neonatal screening is either targeted to high-risk groups, limited to just parts of a country, or is not provided at all.
In the UK, neonatal screening is offered at 5-8 days of age as part of the newborn dried blood spot screening programme for inherited metabolic disorders and cystic fibrosis. The programme is designed to detect all the different forms of clinically important sickle cell disorders: Hb S/S, Hb S/C, Hb S/D-Punjab, Hb S/O-Arab, Hb S/E, and Hb S/β-thalassaemia [15].
Carrier Screening Diagnostics
The key haematological diagnostics for all screening algorithms consist of a full blood count, in particular the determination of the MCV and MCH, the quantification of Hb A2 and Hb F, iron status and screening for the presence or absence of abnormal haemoglobins [21]. Although some other haematological parameters, such the presence of Hb H inclusions and the red cell distribution width (RDW), have proved useful in haemoglobinopathy screening, they are not essential and have not been incorporated into the recommended carrier screening best practice [6].
Many guidelines and diagnostic flow charts for the laboratory diagnosis of haemoglobinopathies have been published. The British Committee for Standards in Haematology, a subgroup of the British Society for Haematology, has published a series of guidelines over the years for UK haematologists and laboratory scientists carrying out haemoglobinopathy screening [22,23] , thalassaemia trait screening [24], foetal DNA analysis [25] and antenatal / newborn screening [26]. The guidelines describe the requirements and approach to screening that are considered practical for the UK population. Although populations have different prevalences of haemoglobinopathies and therefore may require slightly different strategies for haemoglobinopathy screening and the diagnosis of globin gene mutations [27], it has been possible for experts in the field to agree and publish a set of best practice guidelines and an overview of recommended strategies and methods for carrier detection and prenatal diagnosis [6].
There is an important and distinct difference between the algorithms used for carrier screening and those for carrier diagnosis. National carrier screening programmes, such as the UK antenatal screening programme, are not designed to detect all possible haemoglobinopathies in a carrier, due to the costs and resources required in following up too many false negatives with partner testing, and accordingly may have different cut off points to other published schemes. For example, the cut off MCH value of 25pg In the UK diagnostic flow chart, lower than the cut off value of 27pg in the flow chart depicted in Figure 1, is used to reduce the number of α+-thalassaemia investigations and secondly, individuals with a Hb A2 value between 3.0 and 3.5% are considered normal in the UK carrier screening flow chart and are not followed up in order to reduce the costs and workload of partner testing. Thus some antenatal patients with silent/normal Hb A2 β-thalassaemia trait or α+-thalassaemia trait due to a severe type of non-deletion mutation may not be detected by the UK haemoglobinoapthy screening programme flow chart.
A generally accepted flow chart algorithm that achieves an unequivocal carrier diagnosis using the widely accepted screening strategies and cut-off points for the haematological parameters is shown in Figure 1 [28]. The algorithm allows for the detection of carriers with normal Hb A2 β-thalassaemia trait, caused by mutations such as CAP+1 A>C (HBB:c.-50A>C) and IVS1-6 T>C (HBB:c.92+6T>C) , for which some carriers have reduced red cell indices and a HbA2 value between 3.0 and 3.5% [29] (which would be diagnosed as potential alpha thalassaemia trait in a screening algorithm), and for carriers of silent β-thalassaemia mutations such as -101 C>T (HBB:c.-151C>T) and +1480 C>G (HBB:c.*+6C>G) that have both normal red cell indices and a Hb A2 value below 3.0 and 3.5%% [29], often investigated by DNA analysis in a diagnostic case because of a previous diagnosis of a typical β-thalassaemia trait partner or a relation with a mild unexplained β-thalassaemia intermedia phenotype. The algorithm also requires knowledge of other diagnostic pitfalls, such as split HbA2 values due to the presence of a δ-globin chain variant (giving a misdiagnosis of α-thalassaemia trait for a β-thalassaemia carrier), the knowledge that some rare α-chain variants co-migrate with Hb A2, and if expressed at low amounts like Hb Fort Worth (4-5%) it creates a false picture of a high Hb A2 value 6-9% (giving a misdiagnosis of β-thalassaemia trait) [29]. Similarly, Hb pattern analysis requires expertise and knowledge of potential pitfalls, such as understanding that the co-inheritance of an α-chain variant and a β-chain variant creates a new hybrid variant with a different characteristic retention time and electrophoretic mobility to the parent abnormal haemoglobins, creating a complex and often confusing Hb pattern and making diagnostic interpretation difficult [28].
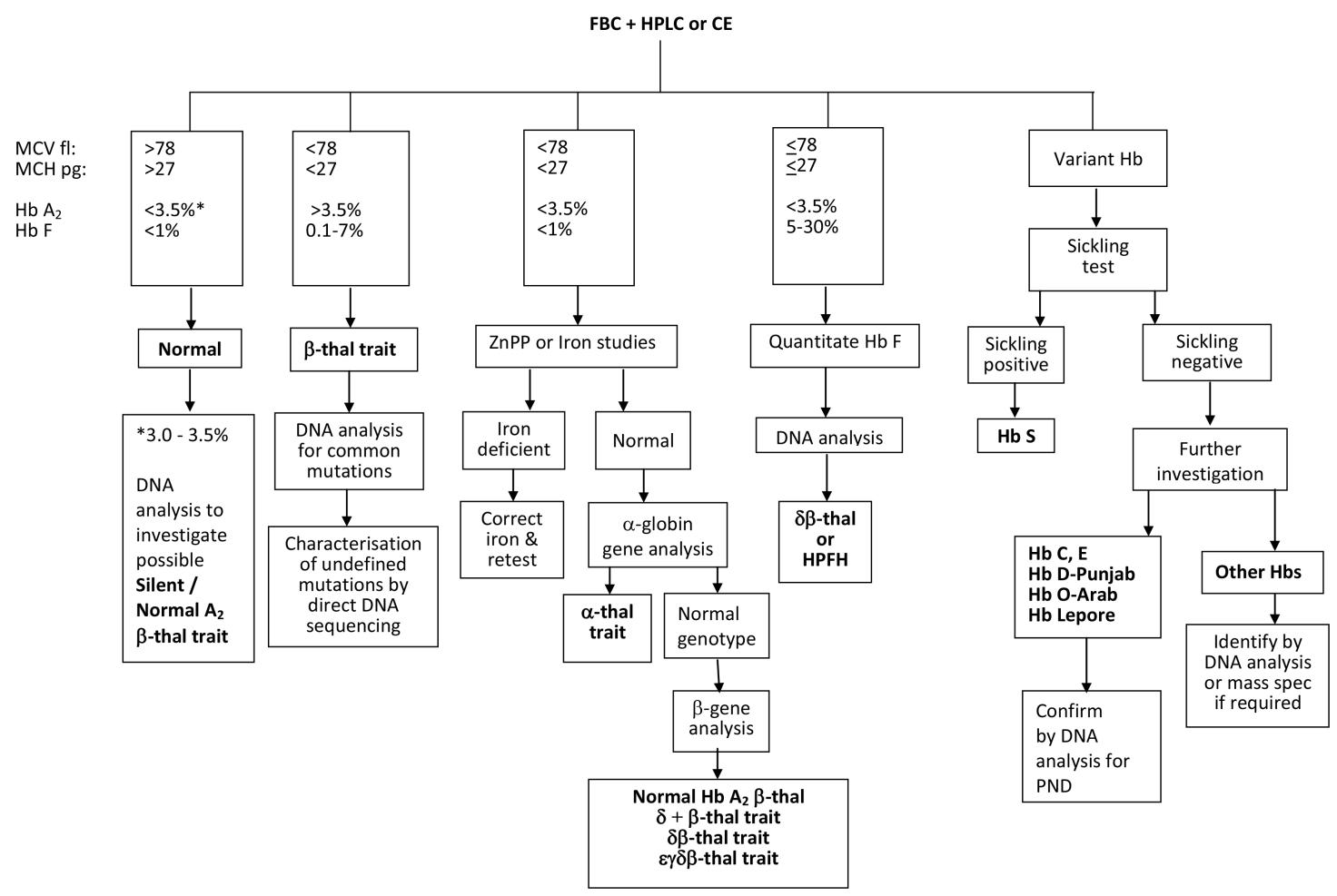
Figure 1 A flowchart for haemoglobinopathy carrier diagnosis using recommended best practice cut off values [6].
Abbreviations: MCV, mean corpuscular volume; MCH, mean cell haemoglobin; Hb, haemoglobin; thal, thalassaemia; ZnPP, zinc protoporphyrin; HPFH, hereditary persistence of fetal haemoglobin; PND, prenatal diagnosis.
Red Blood Cell Indices
The red blood cell indices are usually determined by automated electronic cell counters, with the MCV and MCH being variably reduced in thalassaemia carriers. However the first method used for the screening of thalassaemia was the osmotic fragility test, a fast and simple method which is still used in some low resource countries to screen populations in a modernized form called the Naked Eye Single Tube Redcell Osmotic Fragility Test (NESTROF) [29].
Hb Quantification
The most widely used methods for Hb A2 measurement are high-performance liquid chromatography (HPLC), and less frequently, microchromatography using DEAE cellulose (DE-52). However the newer approach of capillary electrophoresis (CE) is fast, accurate and is becoming fashionable and well established. Both HPLC and CE accurately measure the Hb A2 and Hb F quantities in a similar manner. Isoelectic focusing has proved accurate but is time-consuming and is rarely used nowadays. Another earlier approach, densitometry scanning after cellulose acetate electrophoresis has been proved unsatisfactory and is not recommended by best practice guidelines [21]. Similarly, Hb F is usually measured by using HPLC or CE.
Hb Pattern Analysis
A number of methods are in still current use for separating abnormal haemoglobins from Hb A and providing a presumptive diagnosis by its pattern position or elution time. The earliest methods were gel electrophoresis using acid agarose or citrate agar gel and cellulose acetate electrophoresis at PH 8.6. Although band resolution was poor in some cases, the migration patterns are different for each type of electrophoresis permitting a large number of variants to be differentiated. The more recent technique of isoelectric focussing has become widely used because it offers much sharper resolution of the normal and variant Hb fractions. However the majority of diagnostic laboratories use HPLC for both Hb quantitation and Hb variant screening because the systems are automated, operation of the analysers is simple, and the retention times for more than 300 rare alpha and beta chain variants have now been identified [30]. A relatively new development for Hb pattern analysis, automated capillary zone electrophoresis (CE) is a complementary screening technique to HPLC for the routine detection and measurement of haemoglobins and variants [31]. The analyser is becoming popular because CE patterns are simple and easy to read, because unlike HPLC, the method does not separate haemoglobin derivatives such as glycated fractions. Essentially both HPLC and CE provide a presumptive Hb variant diagnosis with few differences in practice. Another recent screening option is the development of an automated capillary iso-electric focussing (cIEF) analyser for haemoglobins, which has been shown to be useful for screening β-thalassaemia and Hb E carriers in Southeast Asia [32].
α-Thalassaemia: Molecular Diagnostic Techniques
The globin genes were the first genes to be mapped and sequenced in the 1970s by Southern blotting and restriction enzyme mapping using purified cDNA probes reverse transcribed from reticulocyte mRNA. That they were the first genes was due to several factors, partly because considerable information was already known from the study of the protein sequence of haemoglobins, because reticulocytes were a good source of relatively pure globin mRNA, and finally the globin genes were very small in size making them very suitable candidates for use in the newly developed methods of recombinant DNA technology [33]. Consequently they have been used as a prototype in the development of most new molecular techniques of mutation diagnosis, and there is now a multitude of PCR-based techniques that are or have been used to detect globin gene mutations, including dot blot analysis, reverse dot blot analysis, the amplification refractory mutation system (ARMS), denaturing gradient gel electrophoresis (DGGE), mutagenically separated polymerase chain reaction, gap-PCR, restriction endonuclease (RE) analysis, real-time PCR, Sanger sequencing, pyrosequencing, multiplex ligation-dependent probe amplification (MLPA) and gene array systems [34,35]. The main molecular diagnostic approaches in common current use for the haemoglobinopathies are summarized in Table 1.
Past Techniques
The discovery that α-thalassaemia was caused by a deletion of the alpha globin genes was made by cDNA/DNA hybridisation studies in 1974, and this enabled the first prenatal diagnosis of a genetic disease (homozygous alpha zero thalassaemia) to be carried out in 1976 using fetal DNA from cultured amniotic fluid cells [36]. However this approach was quickly superseded by Southern blotting and restriction enzyme mapping, which led to the discovery of all the common alpha plus and zero thalassaemia deletion alleles. Using radioactive DNA probes, the homozygous conditions for the three common alpha zero thalassaemia deletion alleles were diagnosed at the DNA level by the absence of normal alpha globin gene sequences, and the carrier states diagnosed by the presence of characteristic abnormal zeta gene fragments. This approach was the standard diagnostic method for ten years until the development of gap-PCR.
Table 1 The principal DNA analysis methods currently used for the diagnosis of haemoglobinopathies.
|
Globin gene disorder |
Diagnostic methods |
|
αo-thalassaemia |
Gap-PCR, MLPA |
|
α+-thalassaemia (deletional) |
Gap-PCR, MLPA |
|
α+-thalassaemia (non-deletional) |
Sequencing, ASO, RE-PCR, pyrosequencing |
|
β-thalassaemia (deletional) |
Gap-PCR, MLPA |
|
β-thalassaemia (non-deletional) |
Sequencing, ASO, RDB, ARMS, RE-PCR, DGGE, RT-PCR |
|
δ-thalassaemia |
Sequencing |
|
δβ-thalassaemia |
Gap-PCR, MLPA |
|
εγδβ-thalassaemia |
MLPA |
|
HPFH (deletional) |
Gap-PCR, MLPA |
|
HPFH (non-deletional) |
Sequencing, ASO, RE-PCR, pyrosequencing |
|
All Hb variants |
Sequencing |
|
Hb S |
ASO, RDB, ARMS, RE-PCR, pyrosequencing |
|
Hb C |
ASO, RDB, ARMS, pyrosequencing |
|
Hb E |
ASO, RDB, ARMS, RE-PCR, pyrosequencing |
|
Hb D-Punjab |
ASO, RDB, ARMS, RE-PCR, Sequencing |
|
Hb O-Arab |
ASO, ARMS, RE-PCR, Sequencing |
|
Hb Lepore |
Gap-PCR, MLPA |
Current Techniques
Gap-PCR. The technique of multiplex gap-PCR is widely applied as a reliable diagnostic test for the common α+-thalassaemia and αo-thalassaemia deletion mutations, but careful application for prenatal diagnosis has been shown necessary due to the possibility of misdiagnosis caused by allele-drop out [37]. The αo-thalassaemia deletions that are routinely diagnosed by gap-PCR in most laboratories are: the --SEA allele, found in Southeast Asian individuals; the --MED and -(α)20.5 alleles found in Mediterranean individuals; the --FIL allele, found in Filipino individuals and finally the --THAI allele, found in Thai individuals. The two α+-thalassaemia deletion mutations routinely diagnosed are 3.7 kb (-α3.7) and the 4.2 kb (-α4.2) singleα-gene deletion mutations [38,39,40,41,42]. Gap-PCR can also be used to define the exact breakpoints of novel α-thalassaemia deletion mutations, as illustrated by the mapping of a novel 6.3kb α+-thalassaemia deletion in a Chinese carrier [43].
MLPA. The majority of the less common αo and α+-thalassaemia mutations cannot be diagnosed by gap-PCR because their breakpoint sequences have never been determined. Instead these deletion mutations are diagnosed by the multiplex ligation-dependent probe amplification assay (MLPA) [44]. This approach is very useful as it permits the identification of the presence of any α-thalassaemia deletion (known and unknown) in a single test [45]. The technique is also used to diagnose triple and quadruple α-globin gene alleles [46]. Unlike gap-PCR, the MLPA technique does not define the exact breakpoints of a deletion and thus does not provide a definitive identification of a deletion mutation, only a “consistent with diagnosis in carrier diagnosis”; however a definitive identification is not usually required for prenatal diagnosis.
Other Approaches. Other approaches have also been developed to provide quick simple, rapid, accurate and cost effective methods of screening for the deletion mutations. These include the use of real-time quantitative PCR analysis for the Southeast Asian αo-thalassaemia deletion in Taiwan [47], real-time gap-PCR and high resolution melting analysis for the Southeast Asian αo-thalassaemia deletion [48], denaturing HPLC for the diagnosis the 4.2 kb α+-thalassaemia deletion gene in Chinese individuals [49], and the use of an oligonucleotide microarray to detect the Southeast Asian αo-thalassaemia deletion and the 3.7kb and 4.2kb α+-thalassaemia deletions [50,51].
The most widely used approach for the diagnosis of non-deletional α+-thalassaemia, (a term describing the point mutations, small deletions or insertions in one of the two α-globin genes), is the selective amplification of each α-globin gene followed by analysis of the PCR product. For the screening of unknown mutations, DNA sequence analysis is the gold standard method [52]. For the diagnosis of known mutations, many other methods have been applied, such as restriction enzyme digestion of amplified product (RE-PCR), reverse dot-blotting, the amplification refractory mutation system (ARMS-PCR) and pyrosequencing. RE-PCR has been used for the diagnosis of the mutation for Hb Constant Spring mutation [53] and also the α2 gene mutations: initiation codon (ATG →ACG) [HBA2:c.2T>C] and the IVS1 donor site 5 base pair deletion (-TGAGG) [HBA2:c.95+2_95+6delTGAGG]. Reverse dot blotting has been applied for diagnosing six Mediterranean α+-thalassaemia point mutations [54], and the amplification refractory mutation system (ARMS) for diagnosing six common Southeast Asian point mutations [55]. In theory any general technique for the direct detection of point mutations such as allele-specific oligonucleotide hybridisation or allele specific priming may be used for the diagnosis of non-deletion α+-thalassaemia mutations. However such strategies have been developed for just a limited range of non-deletion mutations that are found predominantly in a single ethnic group. The closest approach to a comprehensive screening system is the development of a commercially available strip assay using reverse hybridisation to detect two point mutations in the α1-gene and eleven point mutations in the α2-gene [56].
The techniques and diagnostic strategy used to identify α-thalassaemia mutations is largely dependent on the spectrum of mutations observed in the target population. Molecular diagnosis in a multiethnic population usually requires a strategy involving all three current techniques of gap-PCR, MLPA and α-globin gene sequencing [57].
Latest Developments
aCGH. Array comparative genomic hybridization (aCGH) technology is now a proven method for the detailed characterization of rearrangements in the globin gene cluster. Originally developed as a research tool for investigating genomic imbalances in cancer, aCGH has since been used routinely in the genetic diagnostics of copy number variation in a wide variety of disease genes and gene loci [58,59,60,61]. Several publications have highlighted the advantages of this technique for detecting Copy Number Variations (CNVs) over traditional cytogenetic methods. For the identification of gain or loss of copy numbers from the DNA of a patient a comparative co-hybridization of a differentially fluorophore labelled patient and normal control DNA to the same array is performed. The average ratio between the two fluorescent signals for each probe indicates the gain or loss of the patient’s copy number, and if neighbouring probes are involved this is representative for the presence of a deletion or a duplication of part of a gene or genomic region. Therefore the resolution of array CGH is determined by the physical distance between neighbouring probes and the density of probe coverage over the genomic region of interest.
Considerable improvement of the resolution in using aCGH for the detailed analysis of rearrangements was achieved by reducing the probe spacing from 100 to 20 bp or less and extending the oligonucleotide probes to a length up to 60-80 nt, creating an overlap between probes and thus covering the region of interest as ‘tiles on a roof’ [60,61].
Compared to MLPA, array CGH has an increased resolution allowing more accurate breakpoint mapping. However, to characterize the breakpoints of deletions or duplications at the sequence level, the design of oligonucleotide primers in the breakpoint region as close as possible to the breakpoint is still mandatory for PCR and the subsequent sequencing of the breakpoint fragment. Sometimes the selected primers are unsuccessful in amplifying a fragment across the breakpoint because of the involvement of large repetitive elements or inversions/translocations. Array CGH is not able to distinguish these rearrangements and probe design is mainly limited to locus specific unique sequences as repetitive elements may generate non-specific signals. Several publications have shown that aCGH is a useful tool in the characterization of deletion breakpoints in the alpha-globin gene cluster [60,61,62,63,64].
NGS. Next generation sequencing (NGS) has the potential to identify deletions, inversions and translocations and sequence the breakpoints in a single experiment and the technology is nowadays embedded in most genetic laboratories [65]. It is becoming increasingly important for the mutation detection of small DNA sequence changes, mainly because of the relatively short sequence reads, but to a lesser extent for the detection of large deletions or duplications. The enrichment for the target area where deletion/duplication breakpoints are expected increases the success of NGS in determining breakpoint sequences. Besides Whole-Genome Sequencing (WGS), which is still quite costly, target specific bait-capturing RNA probes are used to enrich for possible DNA fragments across the breakpoint to diminish the part of the genome to be sequenced. The major problem for applying NGS in the diagnostic setting is that the bioinformatics of processing sequence reads is still manual and time-consuming. Efforts are being made in developing specialized bioinformatics tools to reach sufficient reading depth and coverage, to recognition discordant sequence reads as deletion/duplication or inversions and to streamline and automate the bioinformatics approach [66].
Genomic DNA is mechanically sheared into smaller fragments of approximately 500 bp, adapter ligated and subsequently hybridised to specific biotinylated RNA baits complementary to unique sequences in the globin clusters. Using streptavidin coated beads a mini-library consisting almost entirely of DNA fragments of the selected globin gene regions is sequenced on MiSeq or HiSeq after amplifying each molecule by using primers complementary to the adapter sequences. By adding patient specific unique sequence tags to the adapter sequence, analysis of more patient samples by pooling is possible. As both strands of each captured DNA fragment is sequenced in opposite direction, dedicated computer software has been developed to align breakpoint spanning sequences to the reference sequence and recognize so-called ‘split reads’ which appear in the alignment process of single reads from either the 5’part or 3’part of the breakpoint. By investigating the direction and location of the split-reads deletion as well as duplication and inversion breakpoints can be identified and characterised to the base pair level. Examples of the rearrangement breakpoints in the alpha-globin gene cluster, solved by NGS are presented in literature recently [67,68].
NGS has not been adopted as a single technology to diagnose all haemoglobinopathy cases. One of the pitfalls for the alpha-globin gene cluster are the numerous repetitive sequences such as the Alu-repeats, for which no specific RNA bait capture probes can be designed. Also the highly homologous HBA1 and HBA2 paralogs, embedded in duplicated stretches of DNA sequence complicate the alignment to the reference sequence. This currently limits the use NGS for the detection of point mutations in the alpha-globin genes until the sequence chemistry allows for longer reads across the region of homology, thus assigning the phase of the mutation to either the alpha2 or alpha1-globin (HBA2 or HBA1) gene correctly [66].
β-Thalassaemia: Molecular Diagnostic Techniques
Although more than 170 different β-thalassaemia mutations have been characterised [2], only approximately 30 mutations are found at a frequency of 1% or greater in at-risk populations. Most mutations are regionally specific and the spectrum of mutations has now been determined for almost all the at-risk populations in each of the four regionally specific areas (Mediterranean countries, Asian-Indian countries, Southeast Asia and sub-Saharan Africa) [69], making it easy to successfully screen for β-thalassaemia mutations in most cases if the ethnic origin of the patient is known. The diagnostic strategy in many diagnostic laboratories screening for a limited mutation spectrum is to a use a simple and cheap PCR based technique that allows the detection of the common mutations simultaneously [70]. Although a bewildering variety of PCR technologies have been developed and applied for screening β-globin gene point mutations, most diagnostic laboratories are still using a simple and cheap technique based on allele-specific oligonucleotide (ASO) hybridisation or allele-specific priming, eg reverse dot-blotting or ARMS [10]. This approach will identify the mutation in more than 90% of cases and then a further screening for the known rare mutations will identify the defect in most of the remaining cases. Mutations remaining unidentified after this second screening are treated as unknown mutations and then characterised by DNA sequencing.
However, in many European laboratories, the impact of migration by different populations with high frequencies of haemoglobinopathies has led to a significantly enlarged range of mutations that need to be screened for in β-thalassaemia carriers, and has necessitated the implementation of a more comprehensive and simpler screening strategy using DNA sequencing as the main screening method in order to detect a large variety of both common and rare mutations quickly for a prenatal diagnosis is required [71].
Past Techniques
Before the development of PCR, β-thalassaemia mutations were diagnosed by Southern blotting and restriction fragment length polymorphism (RFLP) analysis, with the first prenatal diagnosis being reported by the linkage of a specific Sardinian β-thalassaemia mutation to a Bam HI polymorphism in 1980 [72]. As more RFLPS were discovered within the beta globin gene cluster it was discovered that the RFLPS were in linkage disequilibrium and only a limited number of all the possible combinations of linked RFLPs (haplotypes) existed [73]. This enabled the prenatal diagnosis of any β-thalassaemia mutation by linkage analysis if family studies were possible and the β-thalassaemia alleles were associated with a different haplotype to that of the normal alleles [74]. Direct detection of mutations by Southern blotting only became possible following the development of synthetic allele-specific oligonucleotide (ASO) probes [75]. This new technique for the detection and characterization of known β-thalassaemia mutations provided a useful addition to the armoury of Southern blotting techniques, but for only a short time until Southern blotting was replaced by PCR. However the first PCR technique that gained widespread use for the diagnosis of β-thalassaemia mutations was the approach known as dot-blotting, which also utilised radioactive ASO probes [76]. However the usefulness of screening amplified DNA fixed to a nylon membrane was limited by the need for separate hybridisation steps to test for multiple mutations, and the approach was quickly superseded by the development of the reverse dot-blotting technique, in which the mutation specific probes are fixed to a nylon strip [77].
Current Techniques
RDB. Reverse dot-blotting (RDB) was the first-PCR method to gain widespread use and is still used in some labs today. This technique is compatible with the optimum strategy for screening using a two-step hybridisation procedure using a primer panel for common β-thalassaemia mutations, followed by one for the rare mutations, and has been applied to the diagnosis of β-thalassaemia mutations in Mediterranean individuals [78], African-Americans [79] and Thais [80]. The technique is one of the few methods that been developed commercially with some success for the diagnosis of globin gene mutations. Vienna Lab has marketed globin strip assays using allele-specific oligonucleotide probes which reverse-hybridise to biotinylated DNA and which are currently used in several diagnostic laboratories. The assays cover 21 α-thalassaemia mutations and 22 β-thalassaemia mutations, the latter optimised in separate strips for the detection of the common Mediterranean, Middle Eastern and Indian/Southeast Asian mutations.
Oligonucleotide microarrays. The principle of reverse dot blotting has been brought up to date by the development of oligonucleotide microarrays for the simultaneous detection of multiple β-thalassaemia mutations. This approach promises a one-step strategy for the identification of all possible β-globin gene mutations that result in both β-thalassaemia and β-chain variants [81]. Several groups have now published details of a DNA chip platform which has been used to genotype β-thalassaemia carriers and patients [82]. The approach of tagged single-based extension and hybridisation to glass or flow-through arrays has been developed for the detection of 17 β-globin mutations [83] and a similar approach of arrayed primer extension has been used to detect 23 mutations [84]. Bead-based suspension array technology has been shown to be capable of detecting simultaneously 3 deletions of α-thalassaemia, 3 point mutations of α-thalassaemia, and 17 point mutations of β-thalassaemia in Chinese carriers in a single assay [85]. However the development of single array that will identify any one of the several thousand possible DNA sequence changes in the β-globin gene simultaneously is still awaited and may not be a feasible screening option in the low resource setting of some countries.
Primer-specific Amplification. A number of PCR techniques for detecting β-thalassaemia mutations have been developed based on the principle of primer-specific amplification, but the most widely used method still in current usage is known as the amplification refractory mutation system (ARMS) [86]. ARMS primers have been developed to screen for the common β-thalassaemia mutations found in all the main ethnic groups [87], and have been multiplexed to screen for multiple mutations in a single PCR assa [88]. The method provides a quick screening assay that is cheap and does not require high technology or dedicated instruments and remains useful for both mutation screening and prenatal diagnosis [89].
Other Methods. Other methods for diagnosing point mutations: Denaturing gradient gel electrophoresis (DGGE) is an indirect method which has been widely used to characterise β-thalassaemia mutations without any prior knowledge of the molecular defect [90]. DGGE has been used for prenatal diagnosis of β-thalassaemia in India [91] and Greece [92], and remains a useful approach in low resourced countries for screening of mutations [93]. Restriction enzyme PCR (RE-PCR) has only a limited diagnostic role because very few β-thalassaemia mutations create or abolish a restriction endonuclease site, although its use has been widened by the technique of artificially creating a restriction enzyme site which includes the target mutation [94]. The main use of RE-PCR has been for the analysis of β-globin gene haplotypes to determine the origin of globin gene mutations in different ethnic groups [95].
Many other techniques have been applied for the diagnosis of known β-globin gene point mutations. Denaturing high performance liquid chromatography (DHPLC) has been used for the analysis of polymorphic duplexes created by allele-specific priming [96], the analysis of five common Southeast Asian mutations by multiplex minisequencing [97], multiplex primer extension analysis for 10 Taiwanese mutations [98] and the most common Chinese mutations [99], and the screening for 11 commonest Greek mutations [100]. Real-time PCR quantification and melting curve analysis have been used to provide rapid genotyping for a panel of the most frequent Mediterranean mutations [101] and six Lebanese mutations [102]. All provide rapid and accurate genotyping of the common mutations and are in current use as alternative diagnostic approaches for point mutations.
Gap-PCR and MLPA. The current diagnostic strategy to identify large β0-thalassaemia deletion alleles is the same as that for the α0-thalassaemia deletion alleles – a combination of gap-PCR and MLPA. For a definitive diagnosis by gap-PCR, the deletion breakpoint sequence primers have been identified and published for the 290bp, 532bp, 619bp, 1393bp, 1605bp, 3.5kb, 10.3kb and 45kb β0-thalassaemia deletion alleles, plus Hb Lepore [21].
Latest Developments
NGS. The majority of beta-thalassaemia defects are caused by point mutations in the beta-globin gene and thus can potentially be diagnosed using custom panes for sequencing the globin gene coding regions. Custom panels for the next generation sequencing of coding regions were designed to improve the diagnosis of idiopathic erythrocytosis or congenital haemolytic anaemia using an Ion Torrent Personal Genome Machine (Ion Torrent PGM, Thermo Fisher Scientific, S.L.) [103,104]. Gene panels were selected encompassing only the exonic regions or also exon-intron boundaries, intronic regions known to contain point mutations and 3’and 5’untranslated regions of those genes known or expected to be involved in the disease phenotype. Idiopathic erythrocytosis is a rare disorder which occasionally involves mutations in the globin genes to give rise to high oxygen affinity Hb variants. The targeted NGS sequencing panel encompassed 21 genes amongst which the HBA1, HBA2 and HBB genes, and detected four different mutations coding for variants of the beta-globin chain associated with erythrocytosis, the Hb York, Hb Little Rock, Hb San Diego and Hb Potomac [103].
An NGS panel of 40 genes known to be involved in the pathogenesis of Congenital Haemolytic Anemia, one of which was the beta-globin gene were selected for amplification to include exons, exon-intron boundaries, intronic regions known to contain point mutations and 3’and 5’untranslated regions, amongst which the HBB gene (beta-globin gene). After library preparation by multiplex PCR, followed by ligation of bar coded adapters and purification, single molecules were clonally amplified and sequenced using Ion Hi-Q sequencing chemistry on the Ion Torrent PGM. The panel was tested to identify the mutations for both Hb variants and intronic variations causing beta-thalassaemia, such as HBB:c.315G>C (Hb D Punjab), HBB:c.93-21G>A (IVS1-110G>A) and HBB:c.92+6T>C (IVS1-2T>C) [104].
A dedicated NGS assay (using the Ion Torrent PGM) has been designed for the detection of beta-globin mutations only [105]. The assay covered the complete beta-globin gene, including the 5’promoter region, complete exons and introns and the 3’UTR, except for a small intronic region known to be deprived of mutations. The aim was to test the feasibility of NGS using the Ion Torrent platform to screen large cohorts of carriers simultaneously. Approximately 300 carriers and patients were screened reliably in three runs of 90 minutes each, and a coverage of over 400x, which suggests that with the increase of bar coded adapters up to 300 patients could be analysed in a single run reducing running costs considerably [105].
NGS is also used to map breakpoints of deletions in the beta-globin gene cluster, and has allowed the discovery of the first beta-globin gene duplication ever in a female Hb S carrier with a reduced Hb S percentage (14%) [67]. Enrichment of the target region using RNA baits followed by sequencing on the Illumina MiSeq, and interpretation of split-reads lead to the identification of a 147.5 kb duplication containing two beta-genes, one of which carried the Hb S mutation. A second case of beta-gene duplication was reported by Reading et al. and was discovered by MLPA analysis [106].
δβ-Thalassaemia, HPFH and εγδβ-Thalassaemia
Past Techniques. The δβ-thalassaemia, εγδβ-Thalassaemia, HPFH, and Hb Kenya deletion mutations were characterised first by Southern blotting and restriction enzyme mapping, and were identified by characteristic abnormal DNA fragments that spanned the breakpoints of the deletion mutation. This time-consuming technique remained the only molecular diagnostic approach until gap-PCR and then MLPA were introduced.
Current Techniques. Both gap-PCR and MLPA are now used routinely for a molecular diagnosis. As with the other types of thalassaemia deletion alleles, gap-PCR requires a knowledge of the deletion breakpoint sequences, which still remain unknown for some δβ-thalassaemia alleles. Most of the common δβ-thalassaemia and HPFH alleles, including Hb Kenya, can be diagnosed by gap-PCR, and some novel ones have had gap-PCR primers developed for a quick diagnosis following mapping by MLPA, such as a 163kb Italian (εγδβ)0 deletion [107]. Gap-PCR provides a quick and simple genotype screening method for distinguishing the phenotype of HPFH trait from δβ-thalassaemia trait in Asian Indian, African and Mediterranean individuals with raised Hb F levels [108]. MLPA is used to detect rare or novel deletion alleles, but has also proved a useful but expensive first line screening method, especially for defining the spectrum and frequency of β-globin gene cluster deletion mutations in a particular population, as has been demonstrated for the UK and Chinese populations [109,110] .
Hb Variants
Past Techniques. The major discovery that sickle cell gene in West Africans existed in linkage disequilibrium to a Hpa 1 RFLP [111] enabled the first ever prenatal diagnosis to be achieved by Southern blotting and linkage analysis in 1978 [112] and pioneered the way forward for diagnosing by linkage analysis other inherited genetic disorders for which the genes and mutations were unknown at the time, such as haemophilia, Duchenne muscular dystrophy and cystic fibrosis [33]. Then soon after, it was discovered that the sickle cell mutation could be diagnosed directly by digestion with restriction enzyme Dde 1 [113] and since then, detection of the sickle cell mutation has played a central role in the validation of many new molecular diagnostic techniques as they were developed.
Current Techniques. Positive identification of an unkown Hb variant is currently achieved by selective globin gene amplification and Sanger DNA sequencing. However the clinically important Hb variants that are screened for in prevention programmes (Hb S, Hb C, Hb E, Hb D-Punjab and Hb O-Arab) are confirmed by many different simpler PCR-based diagnostic techniques in most haemoglobinopathy molecular diagnostic laboratories, eg ASO hybridisation, ARMS-PCR, RE-PCR [21].
Future Techniques. The clinical application of modern mass spectrometers to identify the amino acid substitution in haemoglobin variants is a growing field of interest. It is now being used by some laboratories in the UK neonatal screening programme as an alternative method to HPLC and IEF to screen for Hb S [114] and also to identify unknown Hb variants [115].
Prenatal Diagnosis by Fetal DNA Analysis
Fetal DNA analysis is performed when a couple, both carrier of haemoglobinopathy and at risk of having a severely affected child, undergo counselling and prenatal diagnosis. There are three procedures for fetal sampling, including chorionic villus sampling (CVS) in the first trimester of pregnancy, amniocentesis from the 15th week of gestation and fetal blood sampling usually after 18-20 weeks.
Past Techniques
Prenatal diagnosis (PND) was first achieved in 1974 by the study of globin chain synthesis in fetal blood, following the development of the technique of fetal blood sampling. This approach was applied for all of the haemoglobinopathies and proved very successful [116]. However, it had the disadvantage of not being possible until the 18th week of pregnancy, which meant a long wait for the mother and, if indicated, a relatively difficult elective abortion. Once the common thalassaemia mutations were characterized, globin chain synthesis was universally replaced by fetal DNA analysis, originally developed using amniotic fluid DNA to avoid the small but higher risk of fetal loss associated with fetal blood sampling. However, as amniocentesis is a second trimester procedure, most diagnostic centres switched quickly to the first-trimester procedure of chorionic villus sampling (CVS) soon after chorionic villus samples were shown to be a better source of fetal DNA for molecular analysis [117].
Current Techniques
The DNA isolated from chorionic villi provides sufficient amounts of high quality to perform prenatal DNA analysis with most of the procedures described previously. The risk of maternal contamination is low as microscopic dissection allows the removal of contaminating maternal deciduas before DNA extraction. With amniocentesis the DNA yield might be limited due to a low amount of cells, which sometimes requires culturing of the amniotic fluid cells for 10 to 14 days. DNA analysis usually involves amplification of the relevant DNA fragment containing the mutation previously determined in the parents with one of the methods described above [6]. However fetal blood sampling has not entirely disappeared as a diagnostic approach, it permits biochemical methods to be used for the analysis of fetal blood, for example, to detect Hb Bart’s hydrops fetalis syndrome by the absence of Hb F on HPLC or CE.
The risk of fetal loss during the invasive prenatal diagnosis, the need for hospitalization and the emotional and physical burden for the pregnant, has initiated a search for noninvasive approaches to determine the fetal genotype. Since the discovery by Dennis Lo [118] that fetal DNA fragments from the placenta can be found in the maternal circulation, steps have been taken to develop strategies to target the presence of disease-causing mutations or SNP’s linked to the disease phenotype of father in the maternal plasma.
Future Techniques
Noninvasive prenatal diagnosis is now available worldwide for fetal sex-determination in high risk pregnancies of X-linked disorders, and for chromosomal aneuploidies like for chromosomes 13, 18 and 21 [118,119,120]. As far as monogenic disorders are concerned the most obvious application is the detection of the paternally inherited autosomal dominant disease allele absent in the mother. In recessive disorders, like thalassaemia or sickle cell disease, the presence of the paternal disease allele doesn’t distinguish between an affected fetus and merely a carrier of the paternal allele, making invasive testing still obligatory. However, the absence of the paternally inherited disease allele when different to the maternal thalassaemia allele, identifies the fetus as not at risk of having the disease, reducing the need of doing invasive testing to approximately half of the cases.
Many methodologies have been described in literature to perform noninvasive tests in maternal plasma, amongst which targeted massive parallel sequencing [121], exclusion of maternal allele using wild type specific peptide-nucleic acid-mediated enriched PCR [122], COLD-PCR and micro-array [123] as an enrichment method for the mutation carrying paternal alleles in maternal circulation, APEX/thalassochip approach based on the detection of paternal SNPs [124,125,126] and MALDI-TOF mass spectrometry [127]. However, translation into routine clinical practice appeared difficult due to lack of sensitivity, low reproducibility, and sensitivity to contamination or because procedures appear to be too labour intensive [120].
Much progress has been made by developing single molecule analysis technologies to count the individual molecules carrying the mutation or SNPs linked to the disease gene, allowing the determination of the relative mutation dosage (RMD) or relative haplotype dosage (RHDO) respectively. This is a powerful tool not only to determine the paternal contribution to the fetal genotype, but even the maternally inherited mutation to elucidate the fetal mutation status [119].
The relative mutation dosage approach (RMD) makes use of digital PCR (dPCR) and massive parallel sequencing (MPS) counting the single molecular fragments containing the mutation or wildtype sequence from maternal plasma DNA. Slight quantitative alterations are identified of the relative proportions of the wild-type and mutant alleles due to the inheritance of paternal or maternal alleles in addition to the fetal alleles. The rationale lies in the fact that a non-pregnant carrier has a 1:1 ratio of mutant and wildtype allele, however, during pregnancy the relative allele dosage alters due to fetal DNA released in maternal circulation. In the case of a homozygote fetus, the relative dosage would be skewed towards the mutant side, while in the case of a heterozygote fetus the ratio would remain unaltered. Conversely, if the fetus would be normal, the relative dosage is skewed towards the wildtype allele. This methodology has been applied successfully to a case of beta-thalassaemia and a case of sickle cell anaemia [128,129].
The relative haplotype dosage approach (RHDO) measures the balance of maternal haplotypes, similar to the RMD approach, but is applicable to virtually any monogenic disorder. The identification of multiple SNPs assigned to a haplotype makes this approach more robust and as it is no longer limited to the detection of the disease causing mutation itself it can also be used to detect the paternal wildtype allele in maternal circulation to exclude fetal inheritance of the disease causing paternal haplotype.
The single molecule counting by MPS and dPCR is currently considered to be the most robust approach to assess the fetal mutation in maternal plasma [119] RMD and RHDO are the most suitable strategies to determine the fetal inheritance of maternal alleles, and the choice of the one or the other depends on the situation. If the mutations in both parents are known, and haplotype information is lacking the RMD approach is the most straightforward, simple and cost effective method. However, each mutation requires optimization. If on the other hand haplotype information is available then RHDO is a more generic approach with a fixed amount of SNPs forming the haplotype, but is dependent on the informative status of the SNPs present in the paternal alleles being absent in mother. The latter approach will benefit from methods in which a haplotype can be determined instantly [130,131].
Another possible new prenatal diagnosis procedure is DNA analysis of fetal DNA from celomic fluid obtained by colocententesis at 7-9 weeks of gestation after selecting embryo-fetal cells based on their morphology or the use of anti-CD71 MicroBeads. A trial procedure showed concordance between DNA obtained from celomic fluid samples and fetal or newborn DNA in all 302 cases despite maternal contamination of greater thand 60% in approximately 14% of cases [132]. This development provides the opportunity for the use of celocentesis for the early prenatal diagnosis of the haemoglobinopathies [133].
Conclusion
The haematological and molecular techniques used for carrier screening and prevention have evolved considerably since the early pioneering days of the Mediterranean countries setting up programmes for the control of β-thalassaemia and the UK setting up an ad hoc programme of first trimester diagnosis of beta thalassaemia and sickle cell disease in 1982. National antenatal and neonatal screening programmes are now well established in many endemic countries and also in some no-endemic countries because of the growing incidence of immigrant carriers. The widespread adoption of HPLC has much improved the accuracy of screening through more accurate measurement of Hb A2 and Hb F, especially in developing countries with cost pressures such as India. Newer techniques such as capillary electrophoresis have improved the screening for Hb variants. Numerous PCR-based techniques are now used for the molecular diagnosis of carrier mutations, each with their own advantages and disadvantages, and enabling laboratories to choose the ones that meet the needs of their diagnostic mutation repertoire and budget. As carriers are often found to have complex mixture of both alpha and beta globin genotypes which can interact to produce atypical phenotypes, interpretation of the haematological findings and Hb pattern analysis currently plays a key role in the interpretation of the DNA analysis results, by which ever PCR-based method they are generated. In the future, carrier screening by simply sequencing the whole alpha and beta globin gene clusters by next generation sequencing sounds an attractive route to make a diagnosis, as no sequence change should be missed, but the haematology and Hb pattern analysis data will still be needed to be taken into consideration and interpreted together with the sequencing data in order to understand the significance of any observed DNA sequence change from normal. A recently published paper has shown that carrier screening by NGS cab be a competitive screening method to haematological screening methods in populations with a high prevalence of haemoglobinopathies in terms of carrier states detected, but concluded that a correct genetic diagnosis could only be given if the NGS results are interpreted by health-care workers and counsellors properly trained in globin gene genetics and all its clinical manifestations [134].
Currently, most haemoglobinopathy diagnostic laboratories still use in-house protocols for DNA analysis as, probably due to the complexity and heterogeneity of the haemoglobinopathies, few commercial kits for globin gene mutation analysis have been successfully developed and marketed. Similarly, dedicated fully enclosed systems of PCR-based mutation analysis, such as the analysers developed for genotyping red blood cell group antigens, have never materialised. Many of these of PCR techniques will be superseded and replaced in the future by new developments such as next generation sequencing, but the cheapness and simplicity of some of the current techniques will probably ensure their continued use in many diagnostic laboratories worldwide for many years.
Although a lot of the current effort with next generation sequencing technology is being put into target enrichment and disease gene panels, these are just extra steps in improving a diagnostic approach which limits us to small bits of the genome. For the future, the technological way forward in genetic diagnosis is clearly the development of a quick and cheap one-fits-all molecular diagnosis which covers the whole genome sequence of the patient. The software problems are probably greater than the technological ones, for example how to store the patient’s genome data somewhere in digital-space that is protected against viral attacks, and how to develop software that can interpret the NGS-data in an algorithm that asks the right questions. For the haemoglobinopathies, this would have to take into account the patient’s haematological data and Hb separation patterns by HPLC or CE in addition to all of our current knowledge of thalassaemia and Hb variant mutations with their phenotypes.
Acknowledgement
All authors contributed to the data collection and analysis and the writing of this review.
Competing Interests
The authors have declared that no competing interests exist.
References
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization. 2008;86(6):480-7. [CrossRef] [Google scholar] [PubMed]
- Giardine B, Borg J, Viennas E, Pavlidis C, Moradkhani K, Joly P, et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic acids research. 2014;42(Database issue):D1063-9. [CrossRef] [Google scholar] [PubMed]
- Weatherall DJ, Clegg JB. The thalassaemia syndromes: John Wiley & Sons; 2008. [Google scholar]
- Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331-6. [CrossRef] [Google scholar] [PubMed]
- Cao A, Kan YW. The prevention of thalassemia. Cold Spring Harbor perspectives in medicine. 2013;3(2):a011775. [CrossRef] [Google scholar] [PubMed]
- Traeger-Synodinos J, Harteveld CL, Old JM, Petrou M, Galanello R, Giordano P, et al. EMQN Best Practice Guidelines for molecular and haematology methods for carrier identification and prenatal diagnosis of the haemoglobinopathies. European journal of human genetics : EJHG. 2015;23(4):560. [CrossRef] [Google scholar] [PubMed]
- Angastiniotis M, Kyriakidou S, Hadjiminas M. How thalassaemia was controlled in Cyprus. World Health Forum. 1986;7(3):291-7. [Google scholar]
- Cao A, Rosatelli MC, Leoni GB, Tuveri T, Scalas MT, Monni G, et al. Antenatal diagnosis of beta-thalassemia in Sardinia. Annals of the New York Academy of Sciences. 1990;612:215-25. [CrossRef] [Google scholar] [PubMed]
- Loukopoulos D. Haemoglobinopathies in Greece: prevention programme over the past 35 years. The Indian journal of medical research. 2011;134:572-6. [Google scholar]
- Old JM. Screening and genetic diagnosis of haemoglobinopathies. Scandinavian journal of clinical and laboratory investigation. 2007;67(1):71-86. [CrossRef] [Google scholar] [PubMed]
- Community control of hereditary anaemias: memorandum from a WHO meeting. Bulletin of the World Health Organization. 1983;61(1):63-80. [Google scholar] [PubMed]
- Modell B, Organization WH. Guidelines for the control of haemoglobin disorders. 1994. [Google scholar]
- Giordano PC, Harteveld CL, Bakker E. Genetic epidemiology and preventive healthcare in multiethnic societies: the hemoglobinopathies. International journal of environmental research and public health. 2014;11(6):6136-46. [CrossRef] [Google scholar] [PubMed]
- Cousens NE, Gaff CL, Metcalfe SA, Delatycki MB. Carrier screening for beta-thalassaemia: a review of international practice. European journal of human genetics : EJHG. 2010;18(10):1077-83. [CrossRef] [Google scholar] [PubMed]
- Daniel D, Henthorn J. Sickle Cell and Thalassaemia Handbook for Laboratories. NHS Sickle Cell and Thalassaemia Screening Programme. 2012. [Google scholar]
- Therrell BL, Jr., Lloyd-Puryear MA, Eckman JR, Mann MY. Newborn screening for sickle cell diseases in the United States: A review of data spanning 2 decades. Seminars in perinatology. 2015;39(3):238-51. [CrossRef] [Google scholar] [PubMed]
- King L, Knight-Madden J, Reid M. Newborn screening for sickle cell disease in Jamaica: a review - past, present and future. The West Indian medical journal. 2014;63(2):147-50. [CrossRef] [Google scholar] [PubMed]
- Wagner SC, de Castro SM, Gonzalez TP, Santin AP, Zaleski CF, Azevedo LA, et al. Neonatal screening for hemoglobinopathies: results of a public health system in South Brazil. Genetic testing and molecular biomarkers. 2010;14(4):565-9. [CrossRef] [Google scholar] [PubMed]
- Upadhye DS, Jain DL, Trivedi YL, Nadkarni AH, Ghosh K, Colah RB. Neonatal Screening and the Clinical Outcome in Children with Sickle Cell Disease in Central India. PLoS One. 2016;11(1):e0147081. [CrossRef] [Google scholar] [PubMed]
- Bain BJ. Neonatal/newborn haemoglobinopathy screening in Europe and Africa. Journal of clinical pathology. 2009;62(1):53-6. [CrossRef] [Google scholar] [PubMed]
- Old J, Harteveld CL, Traeger-Synodinos J, Petrou M, Angastiniotis M, Galanello R. Prevention of Thalassaemias and Other Haemoglobin Disorders: Volume 2: Laboratory Protocols. Synodinos. 2012. [Google scholar] [PubMed]
- Bain BJ, Amos RJ, Bareford D, Chapman C, Davies SC, Old JM, et al. THE LABORATORY DIAGNOSIS OF HAEMOGLOBINOPATHIES. British journal of haematology. 1998;101(4):783-92. [CrossRef] [Google scholar] [PubMed]
- The British Society for Haematology General Haematology Task Force. Guidelines for haemoglobinopathy screening. Clinical and laboratory haematology. 1988;10(1):87–94. [CrossRef] [Google scholar]
- Guidelines for investigation of the alpha and beta thalassaemia traits. The Thalassaemia Working Party of the BCSH General Haematology Task Force. Journal of clinical pathology. 1994;47(4):289-95. [CrossRef] [Google scholar] [PubMed]
- Guidelines for the fetal diagnosis of globin gene disorders. Globin Gene Disorder Working Party of the BCSH General Haematology Task Force. Journal of clinical pathology. 1994;47(3):199-204. [CrossRef] [Google scholar] [PubMed]
- Ryan K, Bain BJ, Worthington D, James J, Plews D, Mason A, et al. Significant haemoglobinopathies: guidelines for screening and diagnosis. British journal of haematology. 2010;149(1):35-49. [CrossRef] [Google scholar] [PubMed]
- Angastiniotis M, Eleftheriou, A., Galanello, R., Harteveld, C. L., Petrou, M., Traeger-Synodinos, J., Giordano, P., Jauniaux, E., Modell, B., Serour, G. In: Old J, editor. Prevention of Thalassaemias and Other Haemoglobin Disorders: Volume 1: Principles. Nicosia, Cyprus: Thalassaemia International Federation; 2013. [Google scholar]
- J O. Prevention and diagnosis of haemoglobinopathies. A short guide for health professionals and laboratory scientists. Nicosia, Cyprus: Thalassaemia International Federation. 2017. [Google scholar]
- Piplani S, Manan R, Lalit M, Manjari M, Bhasin T, Bawa J. NESTROFT - A Valuable, Cost Effective Screening Test for Beta Thalassemia Trait in North Indian Punjabi Population. Journal of clinical and diagnostic research : JCDR. 2013;7(12):2784-687. [CrossRef] [Google scholar] [PubMed]
- Bain BJ, Wild BJ, Stephens AD, Phelan LA. Variant Haemoglobins: A Guide to Identification2010. [Google scholar]
- Keren DF, Hedstrom D, Gulbranson R, Ou CN, Bak R. Comparison of Sebia Capillarys capillary electrophoresis with the Primus high-pressure liquid chromatography in the evaluation of hemoglobinopathies. American journal of clinical pathology. 2008;130(5):824-31. [CrossRef] [Google scholar] [PubMed]
- Srivorakun H, Fucharoen G, Sanchaisuriya K, Fucharoen S. Diagnosis of common hemoglobinopathies among South East Asian population using capillary isoelectric focusing system. International journal of laboratory hematology. 2017;39(1):101-11. [CrossRef] [Google scholar] [PubMed]
- Wilkie A. Jones EM and Tansey EM (eds): Clinical molecular genetics in the UK c.1975–c.2000. Wellcome witnesses to contemporary medicine, vol 48. Human Genetics. 2016;135(2):257. [Google scholar]
- Old J, Henderson S. Molecular diagnostics for haemoglobinopathies. Expert opinion on medical diagnostics. 2010;4(3):225-40. [CrossRef] [Google scholar] [PubMed]
- Harteveld CL, Kleanthous M, Traeger-Synodinos J. Prenatal diagnosis of hemoglobin disorders: present and future strategies. Clinical biochemistry. 2009;42(18):1767-79. [CrossRef] [Google scholar] [PubMed]
- Kan YW, Golbus MS, Dozy AM. Prenatal diagnosis of alpha-thalassemia. Clinical application of molecular hybridization. The New England journal of medicine. 1976;295(21):1165-7. [CrossRef] [Google scholar] [PubMed]
- Karnpean R, Fucharoen G, Fucharoen S, Sae-ung N, Sanchaisuriya K, Ratanasiri T. Accurate prenatal diagnosis of Hb Bart's hydrops fetalis in daily practice with a double-check PCR system. Acta haematologica. 2009;121(4):227-33. [CrossRef] [Google scholar] [PubMed]
- Dode C, Krishnamoorthy R, Lamb J, Rochette J. Rapid analysis of -alpha 3.7 thalassaemia and alpha alpha alpha anti 3.7 triplication by enzymatic amplification analysis. British journal of haematology. 1993;83(1):105-11. [CrossRef] [Google scholar] [PubMed]
- Bowden DK, Vickers MA, Higgs DR. A PCR-based strategy to detect the common severe determinants of alpha thalassaemia. British journal of haematology. 1992;81(1):104-8. [CrossRef] [Google scholar] [PubMed]
- Baysal E, Huisman TH. Detection of common deletional alpha-thalassemia-2 determinants by PCR. American journal of hematology. 1994;46(3):208-13. [CrossRef] [Google scholar] [PubMed]
- Liu YT, Old JM, Miles K, Fisher CA, Weatherall DJ, Clegg JB. Rapid detection of alpha-thalassaemia deletions and alpha-globin gene triplication by multiplex polymerase chain reactions. British journal of haematology. 2000;108(2):295-9. [CrossRef] [Google scholar] [PubMed]
- Chong SS, Boehm CD, Higgs DR, Cutting GR. Single-tube multiplex-PCR screen for common deletional determinants of alpha-thalassemia. Blood. 2000;95(1):360-2. [Google scholar]
- Wang XY, Lin MX, Lin M. A novel 6.3 kb deletion and the Rare 27.6 kb Deletion Causing alpha+-Thalassemia in two Chinese Patients. Hemoglobin. 2016;40(5):365-8. [CrossRef] [Google scholar] [PubMed]
- Harteveld CL, Voskamp A, Phylipsen M, Akkermans N, den Dunnen JT, White SJ, et al. Nine unknown rearrangements in 16p13.3 and 11p15.4 causing alpha- and beta-thalassaemia characterised by high resolution multiplex ligation-dependent probe amplification. Journal of medical genetics. 2005;42(12):922-31. [CrossRef] [Google scholar] [PubMed]
- Liu JZ, Han H, Schouten JP, Wang LR, Fan XP, Duarte HB, et al. Detection of alpha-thalassemia in China by using multiplex ligation-dependent probe amplification. Hemoglobin. 2008;32(6):561-71. [CrossRef] [Google scholar] [PubMed]
- Harteveld CL, Refaldi C, Cassinerio E, Cappellini MD, Giordano PC. Segmental duplications involving the alpha-globin gene cluster are causing beta-thalassemia intermedia phenotypes in beta-thalassemia heterozygous patients. Blood cells, molecules & diseases. 2008;40(3):312-6. [CrossRef] [Google scholar] [PubMed]
- Sun CF, Lee CH, Cheng SW, Lin MH, Wu TL, Tsao KC, et al. Real-time quantitative PCR analysis for alpha-thalassemia-1 of Southeast Asian type deletion in Taiwan. Clinical genetics. 2001;60(4):305-9. [CrossRef] [Google scholar] [PubMed]
- Pornprasert S, Phusua A, Suanta S, Saetung R, Sanguansermsri T. Detection of alpha-thalassemia-1 Southeast Asian type using real-time gap-PCR with SYBR Green1 and high resolution melting analysis. European journal of haematology. 2008;80(6):510-4. [CrossRef] [Google scholar] [PubMed]
- Ou-Yang H, Hua L, Mo QH, Xu XM. Rapid, accurate genotyping of the common -alpha(4.2) thalassaemia deletion based on the use of denaturing HPLC. Journal of clinical pathology. 2004;57(2):159-63. [CrossRef] [Google scholar] [PubMed]
- Zesong L, Ruijun G, Wen Z. Rapid detection of deletional alpha-thalassemia by an oligonucleotide microarray. American journal of hematology. 2005;80(4):306-8. [CrossRef] [Google scholar] [PubMed]
- Bang-Ce Y, Hongqiong L, Zhuanfong Z, Zhengsong L, Jianling G. Simultaneous detection of alpha-thalassemia and beta-thalassemia by oligonucleotide microarray. Haematologica. 2004;89(8):1010-2. [Google scholar]
- Molchanova TP, Pobedimskaya DD, Postnikov Yu V. A simplified procedure for sequencing amplified DNA containing the alpha 2- or alpha 1-globin gene. Hemoglobin. 1994;18(3):251-5. [CrossRef] [Google scholar] [PubMed]
- Ko TM, Tseng LH, Hsieh FJ, Lee TY. Prenatal diagnosis of Hb H disease due to compound heterozygosity for South-east Asian deletion and Hb constant spring by polymerase chain reaction. Prenatal diagnosis. 1993;13(2):143-6. [CrossRef] [Google scholar] [PubMed]
- Foglietta E, Bianco I, Maggio A, Giambona A. Rapid detection of six common Mediterranean and three non-Mediterranean alpha-thalassemia point mutations by reverse dot blot analysis. American journal of hematology. 2003;74(3):191-5. [CrossRef] [Google scholar] [PubMed]
- Eng B, Patterson M, Walker L, Chui DH, Waye JS. Detection of severe nondeletional alpha-thalassemia mutations using a single-tube multiplex ARMS assay. Genetic testing. 2001;5(4):327-9. [CrossRef] [Google scholar] [PubMed]
- Puehringer H, Najmabadi H, Law HY, Krugluger W, Viprakasit V, Pissard S, et al. Validation of a reverse-hybridization StripAssay for the simultaneous analysis of common alpha-thalassemia point mutations and deletions. Clinical chemistry and laboratory medicine. 2007;45(5):605-10. [CrossRef] [Google scholar] [PubMed]
- Gilad O, Shemer OS, Dgany O, Krasnov T, Nevo M, Noy-Lotan S, et al. Molecular diagnosis of alpha-thalassemia in a multiethnic population. European journal of haematology. 2017;98(6):553-62. [CrossRef] [Google scholar] [PubMed]
- Staaf J, Torngren T, Rambech E, Johansson U, Persson C, Sellberg G, et al. Detection and precise mapping of germline rearrangements in BRCA1, BRCA2, MSH2, and MLH1 using zoom-in array comparative genomic hybridization (aCGH). Human mutation. 2008;29(4):555-64. [CrossRef] [Google scholar] [PubMed]
- Saillour Y, Cossee M, Leturcq F, Vasson A, Beugnet C, Poirier K, et al. Detection of exonic copy-number changes using a highly efficient oligonucleotide-based comparative genomic hybridization-array method. Human mutation. 2008;29(9):1083-90. [CrossRef] [Google scholar] [PubMed]
- Phylipsen M, Chaibunruang A, Vogelaar IP, Balak JR, Schaap RA, Ariyurek Y, et al. Fine-tiling array CGH to improve diagnostics for alpha- and beta-thalassemia rearrangements. Human mutation. 2012;33(1):272-80. [CrossRef] [Google scholar] [PubMed]
- Blattner A, Brunner-Agten S, Ludin K, Hergersberg M, Herklotz R, Huber AR, et al. Detection of germline rearrangements in patients with alpha- and beta-thalassemia using high resolution array CGH. Blood cells, molecules & diseases. 2013;51(1):39-47. [CrossRef] [Google scholar] [PubMed]
- Gilad O, Dgany O, Noy-Lotan S, Krasnov T, Elitzur S, Pissard S, et al. Characterization of two unique alpha-globin gene cluster deletions causing alpha-thalassemia in Israeli Arabs. Hemoglobin. 2014;38(5):319-24. [CrossRef] [Google scholar] [PubMed]
- Jiang H, Liu S, Zhang YL, Wan JH, Li R, Li DZ. Association of an alpha-globin gene cluster duplication and heterozygous beta-thalassemia in a patient with a severe thalassemia syndrome. Hemoglobin. 2015;39(2):102-6. [CrossRef] [Google scholar] [PubMed]
- Liu S, Jiang H, Wu MY, Zhang YL, Li DZ. Thalassemia Intermedia Caused by 16p13.3 Sectional Duplication in a beta-Thalassemia Heterozygous Child. Pediatric hematology and oncology. 2015;32(5):349-53. [CrossRef] [Google scholar] [PubMed]
- Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nature reviews Genetics. 2016;17(6):333-51. [CrossRef] [Google scholar] [PubMed]
- Clark BE, Shooter C, Smith F, Brawand D, Thein SL. Next-generation sequencing as a tool for breakpoint analysis in rearrangements of the globin gene clusters. International journal of laboratory hematology. 2017;39 Suppl 1:111-20. [CrossRef] [Google scholar] [PubMed]
- Shooter C, Rooks H, Thein SL, Clark B. Next generation sequencing identifies a novel rearrangement in the HBB cluster permitting to-the-base characterization. Human mutation. 2015;36(1):142-50. [CrossRef] [Google scholar] [PubMed]
- Clark B, Shooter C, Smith F, Brawand D, Steedman L, Oakley M, et al. Beta thalassaemia intermedia due to co-inheritance of three unique alpha globin cluster duplications characterised by next generation sequencing analysis. British journal of haematology. 2016. [Google scholar]
- Henderson S, Timbs A, McCarthy J, Gallienne A, Van Mourik M, Masters G, et al. Incidence of haemoglobinopathies in various populations - the impact of immigration. Clinical biochemistry. 2009;42(18):1745-56. [CrossRef] [Google scholar] [PubMed]
- Old JM. Screening and genetic diagnosis of haemoglobin disorders. Blood reviews. 2003;17(1):43-53. [CrossRef] [Google scholar] [PubMed]
- Henderson SJ, Timbs AT, McCarthy J, Gallienne AE, Proven M, Rugless MJ, et al. Ten Years of Routine alpha- and beta-Globin Gene Sequencing in UK Hemoglobinopathy Referrals Reveals 60 Novel Mutations. Hemoglobin. 2016;40(2):75-84. [CrossRef] [Google scholar] [PubMed]
- Kan YW, Lee KY, Furbetta M, Angius A, Cao A. Polymorphism of DNA sequence in the beta-globin gene region. Application to prenatal diagnosis of beta 0 thalassemia in Sardinia. The New England journal of medicine. 1980;302(4):185-8. [CrossRef] [Google scholar] [PubMed]
- Orkin SH, Kazazian HH, Jr., Antonarakis SE, Goff SC, Boehm CD, Sexton JP, et al. Linkage of beta-thalassaemia mutations and beta-globin gene polymorphisms with DNA polymorphisms in human beta-globin gene cluster. Nature. 1982;296(5858):627-31. [CrossRef] [Google scholar] [PubMed]
- Old JM, Fitches A, Heath C, Thein SL, Weatherall DJ, Warren R, et al. First-trimester fetal diagnosis for haemoglobinopathies: report on 200 cases. Lancet (London, England). 1986;2(8510):763-7. [CrossRef] [Google scholar] [PubMed]
- Rosatelli C, Falchi AM, Tuveri T, Scalas MT, Di Tucci A, Monni G, et al. Prenatal diagnosis of beta-thalassaemia with the synthetic-oligomer technique. Lancet (London, England). 1985;1(8423):241-3. [CrossRef] [Google scholar] [PubMed]
- Ristaldi MS, Pirastu M, Rosatelli C, Monni G, Erlich H, Saiki R, et al. Prenatal diagnosis of beta-thalassaemia in Mediterranean populations by dot blot analysis with DNA amplification and allele specific oligonucleotide probes. Prenatal diagnosis. 1989;9(9):629-38. [CrossRef] [Google scholar] [PubMed]
- Saiki RK, Walsh PS, Levenson CH, Erlich HA. Genetic analysis of amplified DNA with immobilized sequence-specific oligonucleotide probes. Proc Natl Acad Sci U S A. 1989;86(16):6230-4. [CrossRef] [Google scholar] [PubMed]
- Maggio A, Giambona A, Cai SP, Wall J, Kan YW, Chehab FF. Rapid and simultaneous typing of hemoglobin S, hemoglobin C, and seven Mediterranean beta-thalassemia mutations by covalent reverse dot-blot analysis: application to prenatal diagnosis in Sicily. Blood. 1993;81(1):239-42. [Google scholar]
- Sutcharitchan P, Saiki R, Huisman TH, Kutlar A, McKie V, Erlich H, et al. Reverse dot-blot detection of the African-American beta-thalassemia mutations. Blood. 1995;86(4):1580-5. [Google scholar]
- Sutcharitchan P, Saiki R, Fucharoen S, Winichagoon P, Erlich H, Embury SH. Reverse dot-blot detection of Thai beta-thalassaemia mutations. British journal of haematology. 1995;90(4):809-16. [CrossRef] [Google scholar] [PubMed]
- Cremonesi L, Ferrari M, Giordano PC, Harteveld CL, Kleanthous M, Papasavva T, et al. An overview of current microarray-based human globin gene mutation detection methods. Hemoglobin. 2007;31(3):289-311. [CrossRef] [Google scholar] [PubMed]
- Gemignani F, Perra C, Landi S, Canzian F, Kurg A, Tonisson N, et al. Reliable detection of beta-thalassemia and G6PD mutations by a DNA microarray. Clinical chemistry. 2002;48(11):2051-4. [Google scholar]
- van Moorsel CH, van Wijngaarden EE, Fokkema IF, den Dunnen JT, Roos D, van Zwieten R, et al. beta-Globin mutation detection by tagged single-base extension and hybridization to universal glass and flow-through microarrays. European journal of human genetics : EJHG. 2004;12(7):567-73. [CrossRef] [Google scholar] [PubMed]
- Lu Y, Kham SK, Tan PL, Quah TC, Heng CK, Yeoh AE. Arrayed primer extension: a robust and reliable genotyping platform for the diagnosis of single gene disorders: beta-thalassemia and thiopurine methyltransferase deficiency. Genetic testing. 2005;9(3):212-9. [CrossRef] [Google scholar] [PubMed]
- Yin A, Zhang L, Luo M, He T, Zhang Y, Liu C, et al. Development of bead-based suspension array technology for the diagnosis of thalassemia. American journal of hematology. 2014;89(12):1158-9. [CrossRef] [Google scholar] [PubMed]
- Old JM, Varawalla NY, Weatherall DJ. Rapid detection and prenatal diagnosis of beta-thalassaemia: studies in Indian and Cypriot populations in the UK. Lancet (London, England). 1990;336(8719):834-7. [CrossRef] [Google scholar] [PubMed]
- Old JM, Khan SN, Verma I, Fucharoen S, Kleanthous M, Ioannou P, et al. A multi-center study in order to further define the molecular basis of beta-thalassemia in Thailand, Pakistan, Sri Lanka, Mauritius, Syria, and India, and to develop a simple molecular diagnostic strategy by amplification refractory mutation system-polymer. Hemoglobin. 2001;25(4):397. [CrossRef] [Google scholar] [PubMed]
- Baig SM. Molecular diagnosis of beta-thalassemia by multiplex ARMS-PCR: a cost effective method for developing countries like Pakistan. Prenatal diagnosis. 2007;27(6):580-1. [CrossRef] [Google scholar] [PubMed]
- Miri-Moghaddam E, Bahrami S, Naderi M, Bazi A, Karimipoor M. Molecular Characterization of beta-Thalassemia Intermedia in Southeast Iran. Hemoglobin. 2016;40(3):173-8. [CrossRef] [Google scholar] [PubMed]
- Losekoot M, Fodde R, Harteveld CL, van Heeren H, Giordano PC, Bernini LF. Denaturing gradient gel electrophoresis and direct sequencing of PCR amplified genomic DNA: a rapid and reliable diagnostic approach to beta thalassaemia. British journal of haematology. 1990;76(2):269-74. [CrossRef] [Google scholar] [PubMed]
- Gorakshakar AC, Lulla CP, Nadkarni AH, Pawar AR, Desai SN, Colah RB, et al. Prenatal diagnosis of beta-thalassemia among Indians using denaturing gradient gel electrophoresis. Hemoglobin. 1997;21(5):421-35. [CrossRef] [Google scholar] [PubMed]
- Kanavakis E, Traeger-Synodinos J, Vrettou C, Maragoudaki E, Tzetis M, Kattamis C. Prenatal diagnosis of the thalassaemia syndromes by rapid DNA analytical methods. Molecular human reproduction. 1997;3(6):523-8. [CrossRef] [Google scholar] [PubMed]
- Akhavan-Niaki H, Banihashemi A, Azizi M. Beta globin frameworks in thalassemia major patients from north iran. Iranian journal of pediatrics. 2012;22(3):297-302. [Google scholar]
- Lindeman R, Hu SP, Volpato F, Trent RJ. Polymerase chain reaction (PCR) mutagenesis enabling rapid non-radioactive detection of common beta-thalassaemia mutations in Mediterraneans. British journal of haematology. 1991;78(1):100-4. [CrossRef] [Google scholar] [PubMed]
- Varawalla NY, Fitches AC, Old JM. Analysis of beta-globin gene haplotypes in Asian Indians: origin and spread of beta-thalassaemia on the Indian subcontinent. Hum Genet. 1992;90(4):443-9. [CrossRef] [Google scholar] [PubMed]
- Webster MT, Wells RS, Clegg JB. Analysis of variation in the human beta-globin gene cluster using a novel DHPLC technique. Mutation research. 2002;501(1-2):99-103. [CrossRef] [Google scholar] [PubMed]
- Yip SP, Pun SF, Leung KH, Lee SY. Rapid, simultaneous genotyping of five common Southeast Asian beta-thalassemia mutations by multiplex minisequencing and denaturing HPLC. Clinical chemistry. 2003;49(10):1656-9. [CrossRef] [Google scholar] [PubMed]
- Su YN, Lee CN, Hung CC, Chen CA, Cheng WF, Tsao PN, et al. Rapid detection of beta-globin gene (HBB) mutations coupling heteroduplex and primer-extension analysis by DHPLC. Human mutation. 2003;22(4):326-36. [CrossRef] [Google scholar] [PubMed]
- Wu G, Hua L, Zhu J, Mo QH, Xu XM. Rapid, accurate genotyping of beta-thalassaemia mutations using a novel multiplex primer extension/denaturing high-performance liquid chromatography assay. British journal of haematology. 2003;122(2):311-6. [CrossRef] [Google scholar] [PubMed]
- Bournazos SN, Tserga A, Patrinos GP, Papadakis MN. A versatile denaturing HPLC approach for human beta-globin gene mutation screening. American journal of hematology. 2007;82(2):168-70. [CrossRef] [Google scholar] [PubMed]
- Vrettou C, Traeger-Synodinos J, Tzetis M, Palmer G, Sofocleous C, Kanavakis E. Real-time PCR for single-cell genotyping in sickle cell and thalassemia syndromes as a rapid, accurate, reliable, and widely applicable protocol for preimplantation genetic diagnosis. Human mutation. 2004;23(5):513-21. [CrossRef] [Google scholar] [PubMed]
- Naja RP, Kaspar H, Shbaklo H, Chakar N, Makhoul NJ, Zalloua PA. Accurate and rapid prenatal diagnosis of the most frequent East Mediterranean beta-thalassemia mutations. American journal of hematology. 2004;75(4):220-4. [CrossRef] [Google scholar] [PubMed]
- Camps C, Petousi N, Bento C, Cario H, Copley RR, McMullin MF, et al. Gene panel sequencing improves the diagnostic work-up of patients with idiopathic erythrocytosis and identifies new mutations. Haematologica. 2016;101(11):1306-18. [CrossRef] [Google scholar] [PubMed]
- Del Orbe Barreto R, Arrizabalaga B, De la Hoz AB, Garcia-Orad A, Tejada MI, Garcia-Ruiz JC, et al. Detection of new pathogenic mutations in patients with congenital haemolytic anaemia using next-generation sequencing. International journal of laboratory hematology. 2016;38(6):629-38. [CrossRef] [Google scholar] [PubMed]
- Hassan SM, Vossen RH, Chessa R, den Dunnen JT, Bakker E, Giordano PC, et al. Molecular diagnostics of the HBB gene in an Omani cohort using bench-top DNA Ion Torrent PGM technology. Blood cells, molecules & diseases. 2014;53(3):133-7. [CrossRef] [Google scholar] [PubMed]
- Reading NS, Shooter C, Song J, Miller R, Agarwal A, Lanikova L, et al. Loss of Major DNase I Hypersensitive Sites in Duplicated beta-globin Gene Cluster Incompletely Silences HBB Gene Expression. Human mutation. 2016;37(11):1153-6. [CrossRef] [Google scholar] [PubMed]
- Cardiero G, Prezioso R, Dembech S, Del Vecchio Blanco F, Scarano C, Lacerra G. Identification and molecular characterization of a novel 163 kb deletion: The Italian (gammadeltabeta)(0)-thalassemia. Hematology (Amsterdam, Netherlands). 2016;21(5):317-24. [CrossRef] [Google scholar] [PubMed]
- Craig JE, Barnetson RA, Prior J, Raven JL, Thein SL. Rapid detection of deletions causing delta beta thalassemia and hereditary persistence of fetal hemoglobin by enzymatic amplification. Blood. 1994;83(6):1673-82. [Google scholar]
- Gallienne AE, Dreau HM, McCarthy J, Timbs AT, Hampson JM, Schuh A, et al. Multiplex ligation-dependent probe amplification identification of 17 different beta-globin gene deletions (including four novel mutations) in the UK population. Hemoglobin. 2009;33(6):406-16. [CrossRef] [Google scholar] [PubMed]
- Lee ST, Yoo EH, Kim JY, Kim JW, Ki CS. Multiplex ligation-dependent probe amplification screening of isolated increased HbF levels revealed three cases of novel rearrangements/deletions in the beta-globin gene cluster. British journal of haematology. 2010;148(1):154-60. [CrossRef] [Google scholar] [PubMed]
- Kan YW, Dozy AM. Antenatal diagnosis of sickle-cell anaemia by D.N.A. analysis of amniotic-fluid cells. Lancet (London, England). 1978;2(8096):910-2. [Google scholar]
- Kan YW, Dozy AM. Polymorphism of DNA sequence adjacent to human beta-globin structural gene: relationship to sickle mutation. Proc Natl Acad Sci U S A. 1978;75(11):5631-5. [CrossRef] [Google scholar] [PubMed]
- Chang JC, Kan YW. Antenatal diagnosis of sickle cell anaemia by direct analysis of the sickle mutation. Lancet (London, England). 1981;2(8256):1127-9. [CrossRef] [Google scholar] [PubMed]
- Daniel YA, Henthorn J. Newborn screening for sickling and other haemoglobin disorders using tandem mass spectrometry: A pilot study of methodology in laboratories in England. Journal of medical screening. 2016;23(4):175-8. [CrossRef] [Google scholar] [PubMed]
- Edwards RL, Griffiths P, Bunch J, Cooper HJ. Top-down proteomics and direct surface sampling of neonatal dried blood spots: diagnosis of unknown hemoglobin variants. Journal of the American Society for Mass Spectrometry. 2012;23(11):1921-30. [CrossRef] [Google scholar] [PubMed]
- Alter BP. Antenatal diagnosis. Summary of results. Annals of the New York Academy of Sciences. 1990;612:237-50. [CrossRef] [Google scholar] [PubMed]
- Old JM, Ward RH, Petrou M, Karagozlu F, Modell B, Weatherall DJ. First-trimester fetal diagnosis for haemoglobinopathies: three cases. Lancet (London, England). 1982;2(8313):1413-6. [CrossRef] [Google scholar] [PubMed]
- Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, et al. Presence of fetal DNA in maternal plasma and serum. Lancet (London, England). 1997;350(9076):485-7. [CrossRef] [Google scholar] [PubMed]
- Hudecova I, Chiu RW. Non-invasive prenatal diagnosis of thalassemias using maternal plasma cell free DNA. Best practice & research Clinical obstetrics & gynaecology. 2017;39:63-73. [CrossRef] [Google scholar] [PubMed]
- Ferrari M, Carrera P, Lampasona V, Galbiati S. New trend in non-invasive prenatal diagnosis. Clinica chimica acta; international journal of clinical chemistry. 2015;451(Pt A):9-13. [Google scholar]
- Lam KW, Jiang P, Liao GJ, Chan KC, Leung TY, Chiu RW, et al. Noninvasive prenatal diagnosis of monogenic diseases by targeted massively parallel sequencing of maternal plasma: application to beta-thalassemia. Clinical chemistry. 2012;58(10):1467-75. [CrossRef] [Google scholar] [PubMed]
- Galbiati S, Foglieni B, Travi M, Curcio C, Restagno G, Sbaiz L, et al. Peptide-nucleic acid-mediated enriched polymerase chain reaction as a key point for non-invasive prenatal diagnosis of beta-thalassemia. Haematologica. 2008;93(4):610. [CrossRef] [Google scholar] [PubMed]
- Galbiati S, Monguzzi A, Damin F, Soriani N, Passiu M, Castellani C, et al. COLD-PCR and microarray: two independent highly sensitive approaches allowing the identification of fetal paternally inherited mutations in maternal plasma. Journal of medical genetics. 2016;53(7):481-7. [CrossRef] [Google scholar] [PubMed]
- Papasavva T, Kalakoutis G, Kalikas I, Neokli E, Papacharalambous S, Kyrri A, et al. Noninvasive prenatal diagnostic assay for the detection of beta-thalassemia. Annals of the New York Academy of Sciences. 2006;1075:148-53. [CrossRef] [Google scholar] [PubMed]
- Papasavva T, Kalikas I, Kyrri A, Kleanthous M. Arrayed primer extension for the noninvasive prenatal diagnosis of beta-thalassemia based on detection of single nucleotide polymorphisms. Annals of the New York Academy of Sciences. 2008;1137:302-8. [CrossRef] [Google scholar] [PubMed]
- Papasavva T, van Ijcken WF, Kockx CE, van den Hout MC, Kountouris P, Kythreotis L, et al. Next generation sequencing of SNPs for non-invasive prenatal diagnosis: challenges and feasibility as illustrated by an application to beta-thalassaemia. European journal of human genetics : EJHG. 2013;21(12):1403-10. [CrossRef] [Google scholar] [PubMed]
- Li Y, Di Naro E, Vitucci A, Grill S, Zhong XY, Holzgreve W, et al. Size fractionation of cell-free DNA in maternal plasma improves the detection of a paternally inherited beta-thalassemia point mutation by MALDI-TOF mass spectrometry. Fetal diagnosis and therapy. 2009;25(2):246-9. [CrossRef] [Google scholar] [PubMed]
- Lun FM, Tsui NB, Chan KC, Leung TY, Lau TK, Charoenkwan P, et al. Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc Natl Acad Sci U S A. 2008;105(50):19920-5. [CrossRef] [Google scholar] [PubMed]
- Barrett AN, McDonnell TC, Chan KC, Chitty LS. Digital PCR analysis of maternal plasma for noninvasive detection of sickle cell anemia. Clinical chemistry. 2012;58(6):1026-32. [CrossRef] [Google scholar] [PubMed]
- Snyder MW, Adey A, Kitzman JO, Shendure J. Haplotype-resolved genome sequencing: experimental methods and applications. Nature reviews Genetics. 2015;16(6):344-58. [CrossRef] [Google scholar] [PubMed]
- de Vree PJ, de Wit E, Yilmaz M, van de Heijning M, Klous P, Verstegen MJ, et al. Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nature biotechnology. 2014;32(10):1019-25. [CrossRef] [Google scholar] [PubMed]
- Giambona A, Damiani G, Leto F, Jakil C, Renda D, Cigna V, et al. Embryo-fetal erythroid cell selection from celomic fluid allows earlier prenatal diagnosis of hemoglobinopathies. Prenatal diagnosis. 2016;36(4):375-81. [CrossRef] [Google scholar] [PubMed]
- Giambona A, Leto F, Damiani G, Jakil C, Cigna V, Schillaci G, et al. Identification of embryo-fetal cells in celomic fluid using morphological and short-tandem repeats analysis. Prenatal diagnosis. 2016;36(10):973-8. [CrossRef] [Google scholar] [PubMed]
- He J, Song W, Yang J, Lu S, Yuan Y, Guo J, et al. Next-generation sequencing improves thalassemia carrier screening among premarital adults in a high prevalence population: the Dai nationality, China. Genetics in medicine : official journal of the American College of Medical Genetics. 2017. [Google scholar]

