

The Translational Umbrella - A Novel Approach to the Study of the Biological Basis of Mental Health
Bart A. Ellenbroek 1,*![]() , Weiwen Wang 2
, Weiwen Wang 2![]()
-
School of Psychology, Victoria University of Wellington, Kelburn, Wellington, New Zealand
-
CAS Key Laboratory of Mental Health, Institute of Psychology, Beijing, China
* Correspondence: Bart Ellenbroek ![]()
Received: March 20, 2018 | Accepted: March 21, 2018 | Published: March 29, 2018
OBM Neurobiology 2018, Volume 2, Issue 1, doi:10.21926/obm.neurobiol.1801008
Recommended citation: Ellenbroek B, A. Wang WW. The Translational Umbrella - A Novel Approach to the Study of the Biological Basis of Mental Health. OBM Neurobiology 2018; 2(1): 008; doi:10.21926/obm.neurobiol.1801008.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Keywords
Animal Modelling; Psychiatric Disorders; Etiology; Translational Validity; Biomarker; Endophenotypes
1. Introduction
One in every three individuals in the world will be diagnosed with a brain disorder in their lifetime [1] and, as most of these disorders develop relatively early in life, the impact on patients, family and society as a whole, is tremendous. The Global Burden of Disease study conducted by the World Health Organization estimated that as a group, brain disorders represent by far the greatest burden in terms of years lived with disability [2]. This high burden also translates into enormous costs to society. A study in Europe estimated the annual costs at 800 billion Euro [3], more than the costs associated with lung diseases, diabetes, cardiovascular disorders and cancer combined.
Multiple factors account for these high costs, including the previously mentioned early age of onset of most disorders. Anxiety disorders, major depression, bipolar disorders and schizophrenia typically develop before the age of 30, thus hitting patients in their prime and in the early stages of their career. This aspect of brain disorders, combined with their significant impact on the functioning of patients, explains why the indirect costs (including loss of productivity, social benefits, and early retirement) in particular are so high [3]. Another major contributor to the high costs, both personal and financial, is the limited efficacy of current therapies. Around 30% to 50% of all patients suffering from mood disorders or schizophrenia do not respond satisfactorily to current pharmacological treatments [4,5]. Moreover, even among the responders, it is many weeks before antidepressants induce a meaningful reduction in symptoms and antipsychotics are only effective in treating the positive but not the negative symptoms [6,7]. In addition, no drugs have yet been approved for disorders such as autism spectrum disorders.
Taken together, these data paint a gloomy picture of the prospects for psychiatric patients and emphasize the major unmet medical need. Astonishingly and in spite of this, many major pharmaceutical companies, such as Novartis, GlaxoSmithKline and AstraZeneca, have either fully withdrawn or substantially reduced their research efforts in neuroscience over the last decade [8,9], with Pfizer as the most recent example [10]. This can be primarily attributed to the very poor success rate of drug development in the neuroscience field. In 2004, a landmark study showed that of all the drugs entered into phase 1 clinical trials, only eight eventually made it to the market in the field of neuroscience [11]; this is the lowest rate of all therapeutic areas (together with oncology). More recent studies have indicated that, if anything, this situation has deteriorated [12]. This latter analysis also showed that in the vast majority of cases, drugs fail because of lack of efficacy, rather than pharmacokinetic problems or an unacceptable side-effect profile. Most drugs are generally first tested for efficacy in animal models, suggesting that a flaw in our animal modeling may play a substantial role in the current crisis in the neuroscience drug development field. This was recently illustrated in the analysis of AZD8529, a novel mGluR2 modulator that was developed for the treatment of schizophrenia by AstraZeneca [13]. Phase 2 clinical trials with over 150 patients failed to show an effect that was superior to that of the placebo. Nonetheless, the drug was shown to be effective in seven animal models of schizophrenia.
2. The Limits of the Current Generation of Animal Models
The experience with AZD8529 is certainly not an isolated case and indeed, it has been argued that the lack of any major breakthrough in the treatment of psychiatric disorders is linked to the failure of the current generation of animal models [14,15]. The reasons for this are, in part, related to the basic premise of psychiatric diagnosis itself, and partly due to the failure to incorporate relevant clinical findings into animal models. For many years, the categorical approach to classifying disorders according to the Diagnostic and Statistical Manual (DSM) and the International Classification of Diseases (ICD) has been considered a hindrance in studying the neurobiology (and, by extension, in the development of new drugs) of psychiatric disorders [16,17,18]. The main argument for this is that a diagnosis is assigned according to the DSM and ICD criteria on the basis of having several symptoms from a large checklist. For instance, a diagnosis of schizophrenia based on DSM V requires the presence of any two out of a list of five symptoms; whether a patient has symptoms 1 and 2 or 3 and 5 is irrelevant and both receive the same diagnosis. Thus, by definition, patients diagnosed in this way form a heterogeneous group, likely with different etiologies and pathologies. This is even more clearly illustrated in the case of major depression, in which symptoms include weight loss or weight gain, psychomotor retardation or psychomotor agitation and insomnia or hypersomnia [19]. Consequently, patients with the same diagnosis can have diametrically opposite symptoms, a phenomenon that is impossible to incorporate into an animal model [14] and, seems contradictory from a neurobiological point of view. In addition, it is generally recognized that many psychiatric symptoms occur in multiple disorders [20,21]. For instance, deficits in social cognition are found in autism spectrum disorders, social phobia and schizophrenia among other conditions. Similarly, deficits in anticipatory pleasure have been reported in patients with schizophrenia, major depression and bipolar disorders. These commonalities extend beyond the traditional symptoms and include deficits in cognition (such as working memory and executive functioning), sensory processing (such as prepulse inhibition [22]) and physiological changes (such as reductions in heart rate variability [18]). In recent years, increasing evidence has also shown commonalities in brain pathology [23] and genetic vulnerability factors [24] across psychiatric disorders. In summary, while patients in a single psychiatric disorder group can show different symptoms, patients with different diagnoses may share similar symptoms. Therefore, it seems pertinent to focus more on modeling homogeneous symptoms than on heterogeneous disorders.
Failure to incorporate relevant clinical information further limits the translational validity of our current generation of animal models. For instance, it is now well established that most disorders and most psychiatric symptoms are polygenic (involving multiple genes) [25], and that non-genetic factors also contribute [26]. To date, gene–environment interactions have not been incorporated into animal models to any great extent. For instance, although more than 70 different genetic animal models have been identified in the field of autism spectrum disorders, [27], virtually all of these models are based on single gene alterations, despite the fact that individual genes rarely contribute more than a few percent to the overall disease liability. Similarly, several environmental models have been developed based on prenatal immune activation or treatment with valproate [28]. Nevertheless, the animals models that combine genetic and environmental risk factors are rare [29].
In addition, analysis of the functional consequences of a gene–environment interaction has often been limited to rodent behavior for which there is no human counterpart. Recently, a task force in the field of cognition in schizophrenia tried to identify cognitive domains that have both a human and an animal counterpart [30]. Furthermore, we have recently proposed heart rate variability as a trans-species biomarker of cognitive and emotional flexibility [31].
3. The Translational Umbrella
The previous discussion makes it clear that a change in our current approach to drug discovery is required to substantially improve our chances of successfully developing novel drugs. Therefore, we propose “the translational umbrella” as a metaphor for the optimization of the translational validity between animal and human research based on searching for similarities in etiology, symptoms, physiology and pathology. Moreover, focusing on symptoms rather than disorders is likely to lead to an increase in homogeneity within the models, thereby enhancing the chances of finding meaningful targets and therapies.
Figure 1 provides a schematic representation of “the translational umbrella” approach using major depression as an example. In this example, we focus particularly on pleasure perception, one of the core symptoms of major depression. As discussed previously, all psychiatric disorders have both genetic and environmental components to their etiologies, which must be included under the umbrella. Although acknowledging that major depression is polygenic in nature, a first approximation could focus on a common genetic variant such as the 5-hydroxytryptamine transporter-linked polymorphic region (5-HTTLPR), which comprises a deletion (short, s)/insertion (long, l) variant in the promotor region of the serotonin transporter [32]. While not unequivocal, there is ample evidence linking the s-allele to major depression, a finding that is supported by meta-analyses [33]. More importantly, the s-allele seems to interact with childhood adversity [34], an association that was confirmed in a recent meta-analysis including over 55,000 participants [35]. The relationship between this gene–environment interaction and pleasure perception remains to be investigated, although some supporting preliminary data have been reported [36]. It is becoming increasingly apparent that emotions and cognitive processes are closely related, and even share a common neurobiological substrate, involving the prefrontal/anterior cingulate cortex and the amygdala as well as other networks [37]. Again, while more clinical research is needed to confirm the relationship between pleasure perception and executive functioning, a recent study clearly linked increased pleasure to more efficient executive functioning in patients with major depression [38]. Intriguingly, the presence of the s-allele of the 5-HTTLPR in combination with increased childhood adversity was found to be associated with reduced executive functioning [39], thus linking together the top four levels of the translational umbrella. Moreover, the s-allele of the 5-HTTLPR has often been linked to deficits in the prefrontal cortex/amygdala network, which is involved in both executive functioning and pleasure perception [40]. Heart rate variability is another characteristic that is linked both to emotional and cognitive functioning, as well as to the prefrontal/anterior cingulate cortex-amygdala circuitry [18,31,41,42]. Moreover, major depression is associated with a reduction in heart rate variability [43]. This is particularly interesting as the same methods can be used to measure heart rate variability in both humans and animals [31], making it a perfect parameter for inclusion in the translational umbrella approach. After having laid down the foundation of the translational umbrella in terms of etiology, symptoms, cognition and physiology, we can then use the model to detect biomarkers, which then can help us elucidate the neurobiological mechanisms and identify novel potential drug targets. These biomarkers can be changes in neurotransmitters or brain connectivity patterns, but can also include immunological or endocrine changes.
4. Summary and Outlook
In this editorial, we have proposed the translational umbrella as a novel approach to the study of the neurobiology of mental health. By focusing on symptoms rather than disorders and by studying animals and humans using similar methods, we hope that this approach can provide a way out of the current crisis faced by the pharmaceutical industry. A crucial aspect of this model is the needs for a combined animal/human approach, both of which have so far focused too much on diagnostic “entities” and too little on specific symptoms. As we showed in the major depression example (Figure 1), while gene-environment interaction are critically involved in the etiology of psychiatric disorders, their role in determining specific symptoms is less well understood. Thus, a substantial effort is required to assess whether these interactions are related to specific symptoms, and whether this is valid across diagnoses. Similarly, a major effort is required to develop trans-species markers, such as prepulse inhibition, and heart rate variability, to ensure the success of the translational umbrella approach to drug discovery. Nevertheless, if successful, this approach may help restore faith in the predictive validity of animal models for psychiatric disorders, and increase pharmaceutical efforts in this area once again.
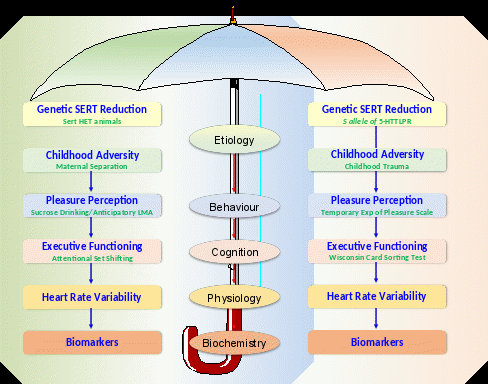
Figure 1: The translational umbrella
Author Contributions
Both authors contributed to writing of this manuscript.
Funding
This editorial was written without any specific funding.
Competing Interests
The authors have declared that no competing interests exist.
References
- Andlin-Sobocki P, Jonsson B, Wittchen HU, Olesen J. Cost of disorders of the brain in Europe. Eur J Neurol. 2005;12:1-27. [CrossRef] [Google scholar] [PubMed]
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2163-2196. [CrossRef] [Google scholar] [PubMed]
- Gustavsson A, Svensson M, Jacobi F, Allgulander C, Alonso J, Beghi E, et al. Cost of disorders of the brain in Europe 2010. Eur neuropsychopharm: the journal of the European College of Neuropsychopharmacology. 2011;21:718-779. [CrossRef] [Google scholar] [PubMed]
- Hendrie C, Pickles A. The failure of the antidepressant drug discovery process is systemic. J Psychopharmacol. 2013;27:407-416. [CrossRef] [Google scholar] [PubMed]
- Samara M, Nikolakopoulou A, Salanti G, Leucht S. How many patients with schizophrenia do not respond to antipsychotic drugs? Eur Neuropsychopharm. 2015;25:S520-S521. [CrossRef] [Google scholar]
- Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382:951-962. [CrossRef] [Google scholar] [PubMed]
- Fusar-Poli P, Papanastasiou E, Stahl D, Rocchetti M, Carpenter W, Shergill S, et al. Treatments of Negative Symptoms in Schizophrenia: Meta-Analysis of 168 Randomized Placebo-Controlled Trials. Schizophr Bull. 2015;41:892-899. [CrossRef] [Google scholar] [PubMed]
- Miller G. Is pharma running out of brainy ideas? Sci. 2010;329:502-504. [CrossRef] [Google scholar] [PubMed]
- Chandler DJ. Something's got to give: psychiatric disease on the rise and novel drug development on the decline. Drug Discovery Today. 2013;18:202-206. [CrossRef] [Google scholar] [PubMed]
- Mullard A. Pfizer exits neuroscience. Nat Rev Drug Discov. 2018;17:86. [CrossRef] [Google scholar] [PubMed]
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711-715. [CrossRef] [Google scholar] [PubMed]
- Arrowsmith J, Miller P. Trial watch: phase II and phase III attrition rates 2011-2012. Nat Rev Drug Discov. 2013;12:569. [CrossRef] [Google scholar] [PubMed]
- Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G, et al. Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13:419-431. [CrossRef] [Google scholar] [PubMed]
- Kaffman A, Krystal JH. New frontiers in animal research of psychiatric illness. Methods Mol Biol. 2012;829:3-30. [CrossRef] [Google scholar] [PubMed]
- Ellenbroek BA, Youn J. Animal modelling in psychiatry. Gene Environment interactions in psychiatry - Nature, nurture, neuroscience. London: Academic Press; 2016. p. 47-73. [CrossRef] [Google scholar]
- Van Praag HM. Nosologomania: a disorder of psychiatry. World J Biol Psychia. 2000;1:151-158. [CrossRef] [Google scholar] [PubMed]
- Beauchaine TP. Taxometrics and developmental psychopathology. Dev Psychopathol. 2003;15:501-527. [CrossRef] [Google scholar] [PubMed]
- Beauchaine TP, Thayer JF. Heart rate variability as a transdiagnostic biomarker of psychopathology. Int J Psychophysiol. 2015;98:338-350. [CrossRef] [Google scholar] [PubMed]
- APA. Diagnostic and Statistical Manual of mental disorders.Fifth Edition. Arlington VA: American Psychiatric Publishing; 2013. [Google scholar]
- Wigman JT, de Vos S, Wichers M, van Os J, Bartels-Velthuis AA. A Transdiagnostic Network Approach to Psychosis. Schizophr Bull. 2017;43:122-132. [CrossRef] [Google scholar] [PubMed]
- Grisanzio KA, Goldstein-Piekarski AN, Wang MY, Ahmed APR, Samara Z, Williams LM. Transdiagnostic Symptom Clusters and Associations With Brain, Behavior, and Daily Function in Mood, Anxiety, and Trauma Disorders. Jama Psychiatry. 2018;75:201-209. [CrossRef] [Google scholar] [PubMed]
- Kohl S, Heekeren K, Klosterkotter J, Kuhn J. Prepulse inhibition in psychiatric disorders--apart from schizophrenia. J Psychiatr Res. 2013;47:445-452. [CrossRef] [Google scholar] [PubMed]
- Goodkind M, Eickhoff SB, Oathes DJ, Jiang Y, Chang A, Jones-Hagata LB, et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry. 2015;72:305-315. [CrossRef] [Google scholar] [PubMed]
- Gatt JM, Burton KLO, Williams LM, Schofield PR. Specific and common genes implicated across major mental disorders: A review of meta-analysis studies. J Psychiatry Res. 2015;60:1-13. [CrossRef] [Google scholar] [PubMed]
- Bogdan R, Salmeron BJ, Carey CE, Agrawal A, Calhoun VD, Garavan H, et al. Imaging Genetics and Genomics in Psychiatry: A Critical Review of Progress and Potential. Biol Psychiatry. 2017;82:165-175. [CrossRef] [Google scholar] [PubMed]
- Ellenbroek BA, Youn J. Gene environment interactions in psychiatry. Nature, Nurture, Neuroscience London: Academic Press; 2016. 355 p. [Google scholar]
- Banerjee-Basu S, Packer A. SFARI Gene: an evolving database for the autism research community. Dis Model Mech. 2010;3:133-135. [CrossRef] [Google scholar] [PubMed]
- Patterson PH. Modeling autistic features in animals. Pediatr Res. 2011;69:34R-40R. [CrossRef] [Google scholar] [PubMed]
- Ellenbroek BA, August C, Youn J. Does Prenatal Valproate Interact with a Genetic Reduction in the Serotonin Transporter? A Rat Study on Anxiety and Cognition. Front Neurosci. 2016;10:424. [CrossRef] [Google scholar] [PubMed]
- Moore H, Geyer MA, Carter CS, Barch DM. Harnessing cognitive neuroscience to develop new treatments for improving cognition in schizophrenia: CNTRICS selected cognitive paradigms for animal models. Neurosci Biobehav Rev. 2013;37:2087-2091. [CrossRef] [Google scholar] [PubMed]
- Ellenbroek BA, Kidwell M, Colussi-Mas J, Youn J. Heart Rate Variability as a translational biomarker for emotional and cognitive deficits. In: Nomikos GG, editor.2018. [Google scholar]
- Lesch KP, Balling U, Gross J, Strauss K, Wolozin BL, Murphy DL, et al. Organization of the human serotonin transporter gene. J Neural Transm Gen Sect. 1994;95:157-162. [CrossRef] [Google scholar] [PubMed]
- Lopez-Leon S, Janssens ACJW, Ladd AMGZ, Del-Favero J, Claes SJ, Oostra BA, et al. Meta-analyses of genetic studies on major depressive disorder. Mol Psychiatr. 2008;13:772-785. [CrossRef] [Google scholar] [PubMed]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Sci. 2003;301:386-389. [CrossRef] [Google scholar] [PubMed]
- Sharpley CF, Palanisamy SKA, Glyde NS, Dillingham PW, Agnew LL. An update on the interaction between the serotonin transporter promoter variant (5-HTIIPR), stress and depression, plus an exploration of non-confirming findings. Behav Brain Res. 2014;273:89-105. [CrossRef] [Google scholar] [PubMed]
- Nikolova Y, Bogdan R, Pizzagalli DA. Perception of a naturalistic stressor interacts with 5-HTTLPR/rs25531 genotype and gender to impact reward responsiveness. Neuropsychobiology. 2012;65:45-54. [CrossRef] [Google scholar] [PubMed]
- Fleck DE, Welge JA, Eliassen JC, Adler CM, DelBello MP, Strakowski SM. Factor analysis of regional brain activation in bipolar and healthy individuals reveals a consistent modular structure. J Affect Disord. 2018;234:14-19. [CrossRef] [Google scholar] [PubMed]
- Light SN, Bieliauskas LA, Zubieta JK, “Top-Down” Mu-Opioid System Function in Humans: Mu-Opioid Receptors in Ventrolateral Prefrontal Cortex Mediate the Relationship Between Hedonic Tone and Executive Function in Major Depressive Disorder. J Neuropsych Clin N. 2017;29:357-364. [CrossRef] [Google scholar] [PubMed]
- Liu J, Cao F, Li P, Lou F, Lavebratt C. 5-HTTLPR, victimization and ecological executive function of adolescents. Psychiatry Res. 2016;237:55-59. [CrossRef] [Google scholar] [PubMed]
- Madsen MK, Mc Mahon B, Andersen SB, Siebner HR, Knudsen GM, Fisher PM. Threat-related amygdala functional connectivity is associated with 5-HTTLPR genotype and neuroticism. Soc Cogn Affect Neurosci. 2016;11:140-149. [CrossRef] [Google scholar] [PubMed]
- Thayer JF, Ahs F, Fredrikson M, Sollers JJ, Wager TD. A meta-analysis of heart rate variability and neuroimaging studies: Implications for heart rate variability as a marker of stress and health. Neurosci Biobehav Rev. 2012;36:747-756. [CrossRef] [Google scholar] [PubMed]
- Thayer JF, Lane RD. Claude Bernard and the heart-brain connection: further elaboration of a model of neurovisceral integration. Neurosci Biobehav Rev. 2009;33:81-88. [CrossRef] [Google scholar] [PubMed]
- Kidwell M, Ellenbroek BA. Heart and soul: heart rate variability and major depression. Behav Pharmacol. 2018;29:152-164. [CrossRef] [Google scholar] [PubMed]

