

FASD and Brain Development: Perspectives on Where We are and Where We Need to Go
Laurie C. Delatour † ![]() , Hermes H. Yeh †
, Hermes H. Yeh †![]()
Department of Molecular and Systems Biology, Geisel School of Medicine at Dartmouth, One Medical Center Drive, Lebanon, NH 03756, USA
† The authors contributed equally to this work.
* Correspondence: Hermes H. Yeh ![]()
Received: March 14, 2017 | Accepted: March 17, 2017 | Published: March 23, 2017
OBM Neurobiology 2017, Volume 1, Issue 1, doi:10.21926/obm.neurobiol.1701002
Academic Editor: Bart Ellenbroek
Recommended citation: Delatour LC, Yeh HH. FASD and Brain Development: Perspectives on Where We are and Where We Need to Go. OBM Neurobiology 2017;1(1):002; doi:10.21926/obm.neurobiol.1701002.
© 2017 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Maternal consumption of alcohol (ethanol) during pregnancy can lead to life-long neurobehavioral and cognitive abnormalities in the offspring, collectively referred to as Fetal Alcohol Spectrum Disorders (FASD). Prenatal exposure to ethanol is one of the leading causes of non-genetic intellectual disability and FASD is an umbrella category that includes Fetal Alcohol Syndrome, Partial Fetal Alcohol Syndrome, Alcohol Related Birth Defects, and Alcohol Related Neurodevelopmental Disorder. Children with a history of in utero exposure to even moderate levels of ethanol frequently present with varying degrees of a broad assortment of deleterious neurobehavioral and cognitive outcomes. This presents formidable challenges in diagnosing FASD, and therefore in its treatment – on the one hand, the wide range of outcomes is not specific to prenatal exposure to ethanol; on the other hand, the diagnosis, by the very nature of the criteria, is conducted after birth while the primary etiology is clearly embryonic, yet unknown and underexplored. For these and other reasons, targeted and effective treatment options for FASD are lacking or at best, ill defined. The overarching theme driving this perspective is that preclinical investigations on brain development are a prerequisite for advancing our understanding of the embryonic cellular and molecular underpinnings of FASD and its associated abnormalities. In addition, preclinical studies will contribute to identifying therapeutic targets that will complement and broaden the scope of current management of FASD. This will guide the development of data-based strategies for intervention during fetal development, rather than being limited to management in newborns and children when irreversible damage has already been done.
Keywords
Fetal alcohol spectrum disorder; cerebral cortex; radial migration; tangential migration; pyramidal neurons; GABAergic interneurons; Intervention in utero
Maternal Consumption of Ethanol and Prevalence of FASD
The developmental disabilities and birth defects associated with FASD are entirely preventable if women abstained from consuming ethanol during pregnancy. While this statement appears to be intuitively clear, it is in practice more theoretical than realistic. In actuality, despite intense public outreach, awareness, and preventive efforts by local and national organizations, an alarming percentage of women drink during pregnancy; many even binge-drink to risky levels.
The prevalence of drinking among women of child bearing age ranges from 27.6% to 69.3% across the United States, with the prevalence of binge drinking within the same cohort ranging from 10.1% to 26.1% [1]. With approximately 50% of pregnancies in the United Stated unplanned [2], a significant number of pregnant women may be unknowingly putting their children at risk. Furthermore, 1 pregnant woman in 10 in the United States reports alcohol use, and 1 in 33 reports binge drinking in the past 30 days [3]. Binge drinking is particularly dangerous due to exceedingly high blood alcohol concentrations, prolonged exposure to ethanol due to a static rate of ethanol metabolism, and periods of withdrawal [4]. Binge drinking during critical time periods in fetal brain development, which may occur even before a woman is aware that she is pregnant, can lead to microcephaly, disruptions of the corpus callosum, and neuronal loss (For reviews see: [4,5]). Children exposed to ethanol during prenatal development displayed characteristics such as learning difficulties, hyperactivity, impulsivity, and disruptive behaviors in school [4,5,6,7,8,9,10]. The prevalence of FASD was found to be in the range of 2% to 5% in a representative population of children in the United States [11], with the prevalence of Fetal Alcohol Syndrome (FAS), the most serious of the diagnoses falling under FASD, between 0.3 and 1.5 infants per 1,000 live births in certain areas of the United States [12]. Furthermore, this problem is not unique to the United States [13]. Globally, about 10% of pregnant women consume alcohol, and the prevalence of FAS is about 15 per 10,000 people [14]. Preclinical reports pique our awareness that the developing brain is particularly vulnerable in FASD. Despite the staggeringly high prevalence of FASD, the mechanisms for the deleterious effects of prenatal exposure to ethanol on fetal brain development are still not fully understood.
Clinical Presentation of FASD Suggests Broad Cortical Dysfunction
FASD and Deficits in Sensory Processing
The clinical features of FASD suggest broad cortical involvement, but many of these deficits converge on the proper functioning of the somatosensory cortex. The somatosensory cortex is important for the integration of sensory information and communication with other areas of the brain. When this system is functioning properly, the individual can focus his/her attention on important stimuli. Sensory integration is the process by which an individual gathers and organizes information detected by one’s senses, and then formulates the appropriate response [15].
Sensory modulation disorders are characterized by an inability to respond appropriately to sensory information from one’s surroundings and include sensory over-responsivity, under-responsivity, and sensory seeking/craving subtypes [16]. Sensory processing dysfunction is associated with deficits in learning, attention, and motor function in addition to problems with language, hyperactivity, and behavior and emotional regulation [15,17]. These are reflected in characteristics commonly reported in children with FASD such as hyperactivity, attention deficits, restlessness, deficits in executive functions, impulsivity, behavioral problems persisting through adulthood, visual-motor integration, fine-motor coordination, and learning deficits [10,17,18] (for review see [5,8,9]). A representative group of school-age children diagnosed with FASD scored significantly lower on the Short Sensory Profile when compared to age and gender matched controls, illustrating significant deficits in categories of tactile sensitivity, auditory filtering, visual/auditory sensitivity, and underresponsive/seeks sensation [19]. These deficits are also correlated with problems with behavior, rule breaking, emotional regulation, and daily living skills [20,21], which are all examples of adaptive behaviors and highlight the connection between sensory dysfunction and such responses [15,17,19,22].
Adaptive behaviors are age-appropriate behaviors that require an ability to process an array of sensory experiences in one’s environment, and then respond purposefully and appropriately [15,22]. These include following rules at school and work, self-care, safety, and social skills [22,23]. Broadly, in children with prenatal exposure to ethanol, problems were found in communication, social skills, and in performing tasks necessary for daily living [20,23], reflecting deficits in age-appropriate adaptive behaviors. While common to FASD, these features are not specific and are also seen in other sensory modulation disorders such as Attention Deficit Hyperactivity Disorder (ADHD) [24,25], Autism [24,26,27,28], Asperger Syndrome [29], and Fragile X Syndrome [27,30]. Sensory processing dysfunction affects every aspect of daily living from academics to caring for one’s self and thus further research into the developmental causes for these deficits could help inform current work focused on treatment options for the affected individuals.
FASD and Deficits in Motor Skills
Disruptions in primary motor and premotor cortex are also likely, given the clinical finding of impaired fine-motor and gross-motor skills in individuals with FASD [31] (for reviews see [5,8]). Reports of balance issues, tremor, and low muscle tone suggest that the effects are more serious than just a developmental delay in gross-motor skills [32,33]. Ataxia and an increased incidence of cerebral palsy have also been reported in individuals with FASD [32]. Specific deficits in measures of higher order cognitive-motor skills, including hand-eye coordination, have also been noted [31,34]. In an extensive literature review conducted by Bay et al., 2011 [35], they found that while prenatal exposure to ethanol does have a detrimental effect on both fine- and gross-motor function, the specific effects of binge drinking are largely understudied. Nonetheless, disruptions in sensory and motor function are undoubtedly linked and the mechanisms behind these dysfunctions remain to be fully elucidated.
FASD and Deficits in Executive Function
In addition to sensorimotor deficits, children exposed to ethanol during prenatal development show deficits in executive functions as evidenced by tasks requiring, for example, planning, selective inhibition, reasoning, cognitive flexibility, and working memory [36,37]. These all can influence judgment and decision making, leading to increased rule-breaking and impulsive behaviors [37]. The prefrontal cortex mediates executive functions including working memory, selective attention, abstract reasoning, language, sensorimotor integration, and sequencing of activity, in addition to behavioral inhibition [38,39], thus implicating its probable role in FASD. Clearly, the effect of in utero exposure to ethanol is multifaceted, disrupting many different areas of the cortex, and ultimately resulting in a constellation of behavioral, cognitive, and sensory deficits.
Modeling Ethanol Consumption and FASD in Rodents
The use of rodent models has significantly advanced the field of prenatal ethanol research. This is in part due to the diversity of models, which can vary in parameters of ethanol exposure such as timing, method of ethanol delivery, and pattern of ethanol exposure. Furthermore, the propensity to voluntarily consume alcohol varies between different mouse strains [40,41]. Each model has its own strengths and weaknesses but, in aggregate, the ability to customize a paradigm to address a specific physiologically relevant question, in addition to an extensive array of transgenic mouse lines, has facilitated progress in this field.
Timing
Because brain development occurs at different stages throughout gestation, the timing of ethanol exposure will differentially affect brain regions and specific processes. Thus, studies are often designed with a certain central nervous system dysfunction in mind. Furthermore, the period of most rapid growth, known as the “brain growth spurt”, occurs at different times in development in different species [42]. In humans, it begins mid-gestation, while in rodents, it does not occur until right around birth [43]. Therefore, investigations focused on third-trimester development in humans are conducted postnatally in rodents. However, the entire process of brain development is dynamic, with the formation of the cortical plate beginning by the 7th week of gestation in humans [44], which is approximately embryonic days 11 and 13 in mice and rats, respectively [45]. In this light, there is justification for investigating the effects of prenatal exposure to ethanol during earlier time points. Through targeting different stages of development by altering the period of prenatal exposure to ethanol, the mechanism behind specific dysfunctions in the central nervous system can be elucidated.
Route of Ethanol Administration
The various routes of ethanol administration increase the feasibility for studying the effects of prenatal exposure to ethanol in rodents. Ingestion of ethanol by pregnant dams is the most common method [46]. Ethanol can be added to a standard liquid food diet, which is then the sole source of food for the rodent during the experimental time window [46,47]. Ethanol can also be administered through drinking water [48]. In a model first described by Rhodes et al. (2005) [49], with the administration of ethanol-containing water for only a short period during the dark cycle (now known as the Drinking in the Dark (DID) paradigm), pregnant dams reached physiologically significant blood alcohol levels (BAL) overtime more consistently than in a simple two-bottle choice paradigm, where both ethanol and water are available for extended periods of time [50,51].
While the delivery of ethanol in either liquid food or water is the simplest and most cost-effective method of administration, one significant drawback is that the exact amount of ethanol consumed cannot be precisely regulated [46]. The administration of ethanol by oral intubation (gavage) is one way to circumvent this drawback. However, this invasive procedure is decidedly more stressful for the pregnant dam, which may confound the results of prenatal ethanol studies [46]. Ethanol can also be delivered directly through subcutaneous or intraperitoneal injection. While this allows for both precise regulation of dosage and timing of exposure and high BALs, it does not mimic the natural mode of ethanol consumption and thus may not accurately reflect the ramifications of drinking [46]. Ethanol can also be administered as a vapor. This method is relatively simple to perform and less stressful than gavage for the animal while still leading to significant BALs, but like injection, inhalation does not resemble the physiological route of ethanol intake [46]. Overall, there is arguably no perfect rodent model for mimicking human ethanol consumption. Nonetheless, the strategies described above have all contributed important insight into our understanding of the outcomes of prenatal exposure to ethanol.
Pattern of Ethanol Consumption
The pattern of ethanol exposure is a key consideration in choosing a method for delivery. An ingestion method or the vapor method are more commonly used for modeling chronic consumption [46]. However, both the liquid food diet [52] and the DID paradigm [51,53,54] have also been used in binge-type exposure models. While a chronic paradigm is necessary for analyzing the effects of prolonged ethanol exposure on brain development, the National Birth Defects Prevention Study reported that the most common pattern of drinking was ethanol consumption during the first month of pregnancy, followed by abstinence [55]. For many women, this period of drinking is very likely to occur before pregnancy recognition. Thus, there is also a need for short-term ethanol exposure models to study the effects of prenatal exposure limited to the very early stages of development. Taking this view, binge-type exposure models are particularly important, given the prevalence of this pattern of drinking among pregnant women, and the risk of exposing the fetus to high BALs for prolonged periods of time [4].
Corticogenesis and Prenatal Exposure to Ethanol
The influence of a teratogen, such as ethanol, on cortical development can have lasting effects on the health and wellbeing of the unborn child. Cortical development is an intricately regulated process that is marked by precisely timed waves of migrating cells in the dorsal telencephalon. There are two principal modes of migration: the radial migration of primordial glutamatergic pyramidal neurons and the tangential migration of GABAergic inhibitory interneurons (Figure 1A) [56,57].
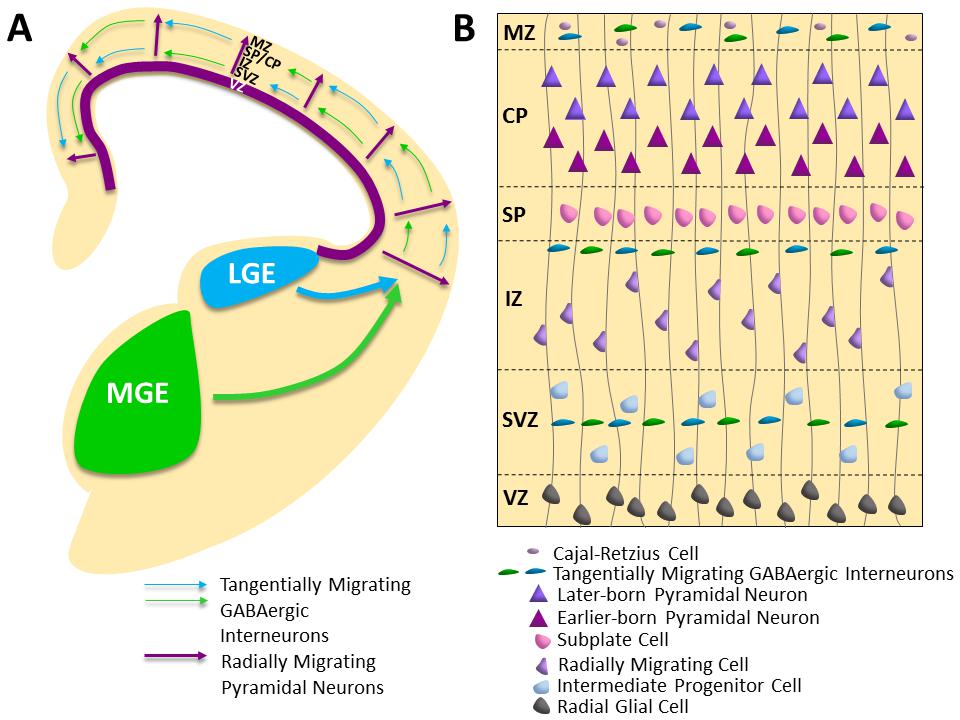
Figure 1 Embryonic Cortical Development in Mouse. (A) Primordial pyramidal neurons migrate radially from the proliferative ventricular zones lining the dorsal telencephalic vesicles (purple) along radial glial fibers. The majority of GABAergic interneurons migrate tangentially into the cortical plate from the medial (MGE; green), lateral (LGE; blue) and caudal (CGE) ganglionic eminences in the ventral telencephalon (CGE not illustrated). (B) Schematic of the embryonic cortical zones at embryonic day 15.5 in the mouse depicting the radial migration of primordial pyramidal neurons along radial glial fibers into the cortical plate (the anlage six layered cortex) and streams of migrating GABAergic interneurons, including a superficial route in the marginal zone and deeper routes through the intermediate and subventricular zones. VZ = Ventricular Zone; SVZ = Subventricular Zone; IZ = Intermediate Zone; SP = Subplate; CP = Cortical Plate; MZ = Marginal Zone.
There are numerous regulatory signals, including the interaction between migrating glutamatergic neurons and GABAergic interneurons, which help to direct corticogenesis [58]. The developing cortex begins as a single layer of neuroepithelial cells and it is through a series of tightly regulated and extremely coordinated processes that a laminar structure with highly complex functional capacities is produced. Therefore, a teratogen, such as ethanol, that disrupts even just one part of this process can have enduring consequences in the central nervous system [52,59,60].
Radial Migration
With glutamatergic pyramidal neurons comprising 70–80% of cortical neurons and providing the sole cortical outflow to subcortical regions and the contralateral cortex [61], the process of radial migration must be tightly regulated to ensure proper cortical development. A snapshot of radial migration at embryonic day 15.5 in the mouse is depicted in Figure 1B. Radial glial cells, functioning as neural stem cells, can divide symmetrically, producing additional radial glial cells, or asymmetrically, resulting in a post-mitotic neuron and an intermediate progenitor cell [62,63]. Intermediate progenitor cells subsequently divide to generate migrating neurons as well [62]. Radial glial and intermediate progenitor cell bodies are found in proliferative zones, delineated as the ventricular zone and subventricular zone, respectively [62,63]. Using the fibers of the radial glial cells as a scaffold, primordial glutamatergic pyramidal neurons migrate radially from the proliferative zones toward the cortical plate, the primitive anlage of the layered cortex, with distinct phases of translocation and locomotion [57,64,65,66]. The first migrating wave of neurons forms the preplate, which is subsequently split into a superficial marginal zone and deeper subplate region by the formation of the cortical plate [56,57,66,67]. The migrating pyramidal neurons stack the cortical plate in an inside-out fashion, such that the earlier born neurons form layers V and VI, and those born later traverse them to form the more superficial layers [57,66,68,69]. An intermediate zone, located between the subventricular zone and cortical plate, contains thalamo-cortical fibers and will become the white matter of the developed cortex [56]. The radial migration of excitatory primordial glutamatergic pyramidal neurons is paired with the tangential migration of inhibitory GABAergic interneurons to establish the intricate balance of excitatory and inhibitory transmission in the developing cortex.
Miller (1988) [70] examined the effects of prenatal exposure to ethanol on regions across the cerebral cortex including sites in motor [71], somatosensory, visual, and auditory cortex. He found that the period of neuron generation was shifted, with a large increase in proliferation later in gestation in ethanol-exposed rats compared to control rats [70]. Furthermore, these later born neurons to a large extent, did not assume their appropriate position in the superficial cortex [70]. There were changes in the size and shape of neurons as well, with earlier born neurons in the ethanol-exposed rats smaller and less eccentric, but more polar, compared to those in age-matched controls, and thus resembling those of immature and migrating neurons [70]. A thinner ventricular zone and thicker subventricular zone in ethanol-exposed rats compared to control rats also reflect this change in proliferation [72]. Therefore, while there is evidence that in utero exposure to ethanol disrupts corticogenesis in the cerebral cortex, the precise mechanism is still unknown and once elucidated, would reveal critical insight into the detrimental effects of ethanol.
Tangential Migration
In contrast to the radial migration of glutamatergic pyramidal neurons, GABAergic inhibitory interneurons migrate tangentially from the ventral telencephalon, primarily from the medial ganglionic eminence (MGE), to the cortex [73] (for reviews see [57,66,74,75]). Once in the cortex, interneurons follow either a superficial or deep migratory stream, through the cortical plate/ marginal zone or subventricular zone/ intermediate zone respectively [57,73]. Towards the end of migration, they adopt a radial migratory pattern as they move into the cortical plate in the developing cortex [56,57,58].
GABA, via activation of GABAA receptors, promotes the tangential migration of primordial GABAergic cortical interneurons [76], which is exacerbated following ethanol exposure in utero [77]. In utero exposure to ethanol affects this process not only through accelerating the migration of GABAergic interneurons but also by significantly increasing the number of GABAergic interneurons in the embryonic neocortex compared to control animals [52,77]. An increase in ambient GABA levels and an upregulation of the expression of GABAA receptors represent prenatal-ethanol-induced extrinsic and intrinsic changes, respectively, that could be influencing abnormal development of GABAergic interneurons [77].
The mechanism behind these changes has been studied using a binge-ethanol exposure paradigm that targets the peak of tangential migration in mice [52]. In utero exposure to ethanol enhanced the migration of GABAergic interneurons from the MGE into the medial prefrontal cortex (mPFC), resulting in both a significant increase in parvalbumin GABAergic interneurons in the mPFC at embryonic day 16.5 and in layer V of the mPFC in postnatal day 70 mice [52]. With the persistent increase in GABAergic interneurons in the young adult mPFC, a commensurate shift in the inhibitory-excitatory balance in layer V pyramidal neurons at postnatal day 70 was characterized by an increase in spontaneous inhibitory post synaptic current frequency (IPSCs) and a reduction in spontaneous excitatory post synaptic current frequency (EPSCs) [52]. An increase in frequency of miniature IPSCs suggested that an increase in the number of inhibitory synapses on mPFC layer V pyramidal neurons could be a possible explanation for the shift in the inhibitory-excitatory balance [52]. The behavioral consequences of these changes at postnatal day 70 were evident by deficits in reversal learning and behavioral flexibility, functions specific to the mPFC [52]. In summary, these experiments provide significant evidence for both the short and long term disruptive effects of binge-type prenatal exposure to ethanol on GABAergic-mediated transmission in the mPFC. Prenatal exposure to chronic ethanol also led to an increase in GABAergic interneurons in the adult mPFC, cortical inhibitory/excitatory imbalance, and hyperactivity, resembling the FASD phenotype in mice [78].
The aberrant prenatal ethanol exposure-induced tangential migration demonstrated by Cuzon et al. (2008) [77] and Skorput et al. (2015) [52] and the previously described ethanol-induced changes in neuronal morphology and time course of neurogenesis described by Miller (1988) [70] are significant because they illustrate that the deleterious effects of prenatal exposure to ethanol are not isolated to just one aspect of cortical development. Rather, they span different brain regions and include both radial and tangential migration.
"Aberrant Migration": A Common Theme in Neurodevelopmental Disorders
In addition to radial and tangential migration, other modes of migration contribute to shaping the immature brain. Aberrant migration is a common theme across many neurodevelopmental disorders. These include schizophrenia, X-linked lissencephaly with abnormal genitalia, autism, and several genetic disorders, among others.
Disruption in the cytoarchitecture and development of layers II, III, and V are evident in the entorhinal cortex of postmortem brains from individuals with schizophrenia [79]. It was found that layer II was almost completely absent of neurons and there was an ectopic band of neurons at the top of layer III [79]. These ectopic neurons were smaller and resembled immature migrating neurons, suggesting a defect in migration [79]. In addition, an altered distribution of neurons was found in the dorsal-lateral prefrontal cortex in the superior frontal gyrus of postmortem brains from individuals with schizophrenia [80]. Specifically, there was an increase in NADPH-d neurons in the deep subcortical white matter, in conjunction with a decrease in NADPH-d neurons in the gray matter and superficial cortical white matter [80]. NADPH-d neurons are a major component of the subplate during development, so their aberrant distribution could reflect a disruption of migration into the subplate or the programmed cell death of these cells during development [80]. These data are consistent with disrupted migration as a possible underlying factor in the pathology of schizophrenia.
Kato and Dobyns (2005) [81] coined the term “developmental interneuronopathy” to describe disorders such as X-linked lissencephaly with abnormal genitalia, which result, in part, from abnormal tangential migration during embryonic development. Patients with this disorder suffer from intractable seizures from the day of birth, developmental delays, chronic diarrhea, and hypothermia [81]. The pathophysiology behind the seizure disorder could be a deficiency in GABAergic interneurons, as seen in both postmortem studies and in a genetic mouse model [81]. This pointed to abnormal tangential migration during embryonic development as an underlying mechanism.
Furthermore, one possible cause for the collection of symptoms associated with autism could be defects in migration and a disruption in neurogenesis [82]. Evidence of dysplasia and heterotopias in the neocortex, dentate gyrus, and cerebellum suggest that altered migration is not specific to one area of the brain [82]. In these areas, distorted neuronal morphology, suggesting poor differentiation, and abnormal laminar distribution were found [82]. Further studies noted disordered lamination, specifically in the anterior cingulate cortex, in postmortem brains from autistic individuals, suggesting aberrant migration within the cortical plate [83]. This led Simms et al. (2009) [83] to postulate that the origin of this defect is early in fetal development.
There are also several genetic disorders that are linked to dysfunctions in neuronal migration. Periventricular heterotopia is an X-linked dominant condition that is only seen in affected females, as males do not survive [84]. In females who are heterozygous for the mutation, random X inactivation leads to one population of cells that migrates correctly into the cortical plate, and another that remains arrested in the ventricular zone [84]. The affected individuals suffer from epilepsy [84]. One of the clinical manifestations of another genetic disorder, lissencephaly type I, is also epilepsy, in addition to severe intellectual deficits [84,85]. It is caused by a mutation in the LIS1 autosomal gene on chromosome 17 that results in absent or malformed gyri and a four-layered cortex due to defects in migration [84]. X-linked lissencephaly is caused by a mutation in doublecortin and results in a similar phenotype in males to lissencephaly type I, and double cortex (subcortical band heterotopia) malformation in heterozygous females [84,85,86,87]. The double cortex phenotype includes a typical six-layered cortex; however, this is in addition to an ectopic population of neurons in the subcortical white matter [84]. On the contrary, lissencephaly type II is due to neuronal over-migration, where neurons migrate past the marginal zone and into the leptomeninges [85]. This results in a “cobblestone” cortex, a thickened gray matter with interrupting bands of white matter, and potentially hydrocephalus [85]. Autosomal recessive Fukuyama Congenital Muscular Dystrophy and Walker-Warburg Syndrome are examples of lissencephaly type II [84,85]. These, in addition to several other known genetic disorders associated with aberrant migration, highlight the significance of proper cortical development in normal brain development and function.
Given the known association of other neurodevelopmental disorders with disruptions in cortical development, it is important to focus on such deficits in light of prenatal exposure to ethanol to better understand the underlying etiology of FASD. In fact, many of the hallmark features of FASD including intellectual and attention deficits, difficulty with word comprehension, sensory processing dysfunction, and impaired visual-motor integration, nonverbal learning, fine-motor skills, and coordination [5,8,17,18,19,22,34] demonstrate that cortical dysfunction is a significant component of FASD. It is likely that it is a combination of aberrant migratory patterns throughout the brain that result in the complexity of symptoms seen in many neurodevelopmental disorders.
Treatment Interventions for FASD: Where We Need to Go
There is no cure for FASD or even any specific treatment. However, early intervention therapies, particularly in the areas of behavior and education, have been shown to be helpful in allowing affected individuals to reach his/her greatest potential [88]. These interventions may include occupational, speech and language, social skills, and physical therapy [88]. Medical treatment for some of the symptoms such as hyperactivity and depression/anxiety may also be a part of the overall treatment plan, however they are not specific to FASD [88]. While these interventions are all beneficial, if they could be coupled or complemented with specific FASD-targeted pharmacological therapies, then the interventions already in place could potentially be even more effective. This requires a better understanding of the mechanisms behind the detrimental effects of prenatal exposure to ethanol.
Early in development, GABA is paradoxically depolarizing [89,90]. Therefore, GABAergic interneurons can form excitatory synapses on neighboring cells. This is largely due to a difference in the intracellular chloride concentration [90]. NKCC1 is a Na+-K+-2Cl- co-transporter that functions to pump sodium, potassium, and chloride ions into cells [91]. On the contrary, KCC2 is a K+-Cl- cotransporter and pumps both ions out of cells [91]. Early in brain development, the balance between NKCC1 and KCC2 expression is tipped in favor of NKCC1, but with maturity, KCC2 expression in the brain ultimately predominates [90–92]. However, before this developmental switch and while NKCC1 expression is higher, there is an increase in the intracellular chloride concentration such that when a ligand binds to and opens the GABAA receptor ionophore, chloride flows out of the cells down its concentration gradient, and thus depolarizes the cell [90,91]. In light of the fact that binge-type in utero exposure to ethanol results in an increase in GABAergic interneurons evident at both embryonic day 16.5 and in young adult mice [52], the depolarizing and therefore potentially excitatory actions of these cells could be part of the underlying cause for prenatal ethanol-induced cortical dysfunction. In testing this mechanistic hypothesis, when the loop diuretic bumetanide has been co-administered with ethanol to pregnant dams, the number of GABAergic interneurons in the mPFC of their offspring is no different from that of the offspring of control dams (Skorput and Yeh, unpublished). This suggests a role for NKCC1 in the inhibitory/excitatory imbalance described by Skorput et al., 2015 [52], and an avenue for future research to further elucidate the mechanism.
Another mode of intervention under investigation is choline supplementation. Choline is not only an essential nutrient, but also a precursor to the neurotransmitter acetylcholine and components of the cell membrane, including phosphatidylcholine and sphingomyelin [93]. It can also serve as a methyl donor [93], highlighting its potential role in epigenetics. Choline administration has proven to be successful in abating some of the behavioral effects of prenatal exposure to ethanol. When ethanol and choline were co-administered from gestational days 5–20 in rats, the offspring exhibited significant improvements in reflex maturation [94], exploratory behavior [95], and spatial working memory [95] compared to ethanol only-exposed offspring. Through altering the timing of choline administration relative to ethanol exposure, further insight regarding its potential therapeutic effects has been garnered. With ethanol exposure from postnatal days 4–9 and choline supplementation from postnatal days 4–30, ethanol only-exposed mice exhibited hyperactivity and an increase in perseverate errors on a spatial reversal learning task, both of which were mitigated by choline administration [96]. In rats, following ethanol exposure from postnatal days 4–9, choline administration from either postnatal days 11–20 or postnatal days 21–30 mitigated the ethanol-induced deficits in spatial memory [97]. The finding that choline has beneficial effects even when administered after exposure to ethanol suggests the exciting prospect of dietary interventions in treating FASD.
The mechanism behind the effects of choline supplementation is yet to be determined, although it is thought that it may be specifically affecting the functioning of the hippocampus and/or prefrontal cortex [95]. In rats exposed to ethanol from postnatal days 4–9, the density of M2/M4 receptors was significantly increased compared to controls in the hippocampus [98]. However, when choline was administered from postnatal days 4–30 in conjunction with ethanol exposure from postnatal days 4–9, there was no longer a significant difference [98]. This is likely just one of many effects of choline supplementation and further investigations in this field will better our understanding of its interaction with prenatal exposure to ethanol.
With preclinical studies in rodents providing strong evidence for the benefits of choline supplementation in mitigating some of the adverse effects of prenatal exposure to ethanol, a randomized, double-blind, placebo-controlled clinical trial was conducted to determine its effectiveness in humans (https://clinicaltrials.gov/ct2/show/NCT01911299). Children ages five through ten with a history of prenatal exposure to ethanol were included in the study, and were randomly assigned to a six-week treatment regimen of 625mg of choline per day, or a placebo [99]. Despite the promising preclinical results, there were no significant improvements observed in learning and memory, executive function, or sustained attention in the choline-treated group compared to the placebo [99]. Investigations in rodents have suggested that the target therapeutic window for choline supplementation may be earlier than the time range used in this study, and thus this is one possible explanation for the unanticipated results [99]. Furthermore, it is possible that the sample size was too small or that the treatment duration was too short to see any significant effects of choline supplementation [99]. Regardless, future studies are necessary to further elucidate the mechanism behind the beneficial effects of choline supplementation in preclinical studies in order to better understand its potential effectiveness in humans.
Peroxisome proliferator-activated receptor (PPAR)-γ is involved in glucose metabolism and adipogenesis, but its role in mediating the inflammatory response in the central nervous system [100] has spurred investigations into its possible therapeutic efficacy in FASD. In both in vitro and in vivo studies, PPAR-γ agonists have been shown to mitigate effects of ethanol-induced toxicity on microglia and neuron loss [101]. Specifically, in a mouse model for FASD, both endogenous and synthetic PPAR-γ agonist administration one day prior to and during perinatal ethanol exposure, significantly decreased the effects of ethanol-induced toxicity in the cerebellum [101]. This included attenuating the loss of microglia and Purkinje cells [101]. Furthermore, when a PPAR-γ agonist was administered one hour prior to ethanol administration to mice during the perinatal period, it mitigated the ethanol-induced increase in production of the mRNA of proinflammatory cytokines and chemokines in the hippocampus, cerebellum, and cerebral cortex [102]. PPAR-γ agonist administration also prevented the change in morphology of microglia cells to a form suggesting activation that was seen following perinatal exposure to ethanol [102]. The mechanism for how PPAR-γ agonists protect microglia and neurons from the damaging effects of ethanol during development is still unknown.
Conclusion
FASD is a complex condition that, while completely preventable, is a leading cause of intellectual disability in the United States. There is evidence that, like other neurodevelopmental disorders, a significant underlying cause for deficiencies in sensory processing, cognition, and behavior is aberrant cortical development. From cell proliferation, to migration, lamination of the cortex, and circuit formation, these are all intricately regulated processes and integral in the proper establishment of the cortical inhibitory/excitatory balance and communication within the cortex and to subcortical areas. When a teratogen is present, it can disrupt the time course and regulation of these events during an extremely formative time for the unborn child, leading to lasting consequences. Therefore, as ongoing FASD research continues to explore fields such as the pathophysiology of and treatment options for the affected individual, an emphasis on embryonic development is crucial to fully understanding and appreciating the complexity of FASD and the design of targeted therapeutic interventions during pregnancy.
Acknowledgments
We thank the following colleagues for comments and critical reading of the manuscript: Bryan W. Luikart, Ph.D., Paul D. Manganiello, M.D., Donald Bartlett Jr., M.D., and Pamela W.L. Yeh.
Author Contributions
LCD and HHY planned the content of the manuscript. LCD wrote the first draft of the manuscript.
Funding
This work was supported in part by PHS NIH grants R01 AA023410 (HHY), R21 AA024036 (HHY), F30 AA025534 (LCD).
Competing Interests
The authors have declared that no competing interests exist.
References
- Center for Disease Control and Prevention. Fetal Alcohol Spectrum Disorders (FASDs): Data & Statistics. Atlanta (GA): Centers for Disease Control and Prevention, National Center on Birth Defects and Developmental Disabilities, Division of Birth Defects and Developmental Disabilities. Available from: https://www.cdc.gov/ncbddd/fasd/data.html (Accessed on January 23, 2017). [PubMed]
- Finer LB, Zolna MR. Unintended pregnancy in the United States: incidence and disparities, 2006. Contraception. 2011;84(5):478-85. [CrossRef] [Google scholar] [PubMed]
- Tan CH, Denny CH, Cheal NE, Sniezek JE, Kanny D. Alcohol use and binge drinking among women of childbearing age-United States, 2011-2013. MMWR Morb Mortal Wkly Rep. 2015;64(37):1042-46. [CrossRef] [Google scholar] [PubMed]
- Maier SE, West JR. Drinking patterns and alcohol-related birth defects. Alcohol Res Health. 2001;25(3):168-74. [Google scholar]
- Riley EP, McGee CL. Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Exp Biol Med. 2005;230(6):357-65. [CrossRef] [Google scholar] [PubMed]
- Behnke M, Smith VC. Committee on Substance Abuse and the Newborn. Prenatal substance abuse: short- and long-term effects on the exposed fetus. Pediatrics. 2013;131:e1009-e1024. [CrossRef] [Google scholar] [PubMed]
- Hoyme HE, Kalberg WO, Led MA, Elliot AJ, Blankership J, Buckley D, et al. Updated clinical guidelines for diagnosing fetal alcohol spectrum disorders. Pediatrics. 2016;138:1-18. [CrossRef] [Google scholar] [PubMed]
- Mattson SN, Riley EP. A review of the neurobehavioral deficits in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol Clin Exp Res. 1998;27(2):279-94. [CrossRef] [Google scholar] [PubMed]
- Sokol RJ, Delaney-Black V, Nordstrom B. Fetal alcohol spectrum disorder. JAMA. 2003;290(22):2996-9. [CrossRef] [Google scholar] [PubMed]
- Streissguth AP, Aase JM, Clarren SK, Randels SP, LaDue RA, Smith DF. Fetal alcohol syndrome in adolescents and adults. JAMA. 1991;265:1961-7. [CrossRef] [Google scholar]
- May PA, Baete A, Russo J, Elliott AJ, Blankenship J, Kalberg WO, et al. Prevalence and characteristics of fetal alcohol spectrum disorders. Pediatrics. 2014;134(5):855-66. [CrossRef] [Google scholar] [PubMed]
- Center for Disease Control and Prevention. Fetal alcohol syndrome- Alaska, Arizona, Colorado, and New York, 1995-1997. MMWR Morb Mortal Wkly Rep. 2002;51(20):433-5. [Google scholar]
- Riley EP, Infante MA, Warren KR. Fetal alcohol spectrum disorders: an overview. Neuropsychol Rev. 2011;21(2):73-80. [CrossRef] [Google scholar] [PubMed]
- Popova S, Lange S, Probst C, Gmel G, Rehm J. Estimation of national, regional, and global prevalence of alcohol use during pregnancy and fetal alcohol syndrome: a systematic review and meta-analysis. Lancet Glob Health. 2017;5(3):e290-e299. [CrossRef] [Google scholar] [PubMed]
- Ayres AJ, Robbins J. Sensory integration and the child: Understanding hidden sensory challenges. Los Angeles: Western Psychological Services; 2005. p.1-p.40. [Google scholar]
- Miller LJ, Anzalone ME, Lane SJ, Cermak SA, Osten ET. Concept evolution in sensory integration: A proposed nosology for diagnosis. Am J Occup Ther. 2007;61(2):135-40. [CrossRef] [Google scholar] [PubMed]
- Franklin L, Deitz J, Jirikowic T, Astley S. Children with fetal alcohol spectrum disorders: problem behaviors and sensory processing. Am J Occup Ther. 2008;62:265-73. [CrossRef] [Google scholar] [PubMed]
- Mattson SN, Riley EP, Delis DC, Jones KL. Neuropsychological comparison of alcohol-exposed children with or without physical features of fetal alcohol syndrome. Neuropsychology. 1998;12(1):146-53. [CrossRef] [Google scholar] [PubMed]
- Jirikowic T, Olson HC, Kartin D. Sensory processing, school performance, and adaptive behavior of young school-age children with fetal alcohol spectrum disorders. Phys Occup Ther Pediatr. 2008;28(2):117-36. [CrossRef] [Google scholar] [PubMed]
- Whaley SE, O’Connor MJ, Gunderson B. Comparison of the adaptive functioning of children prenatally exposed to alcohol to a nonexposed clinical sample. Alcohol Clin Exp Res. 2001;25(7):1018-24. [CrossRef] [Google scholar]
- Streissguth AP, Bookstein FL, Barr HM, Sampson PD, O’Malley K, Young JK. Risk factors for adverse life outcomes in fetal alcohol syndrome and fetal alcohol effects. J Dev Behav Pediatr. 2004;25(4):228-38. [CrossRef] [Google scholar] [PubMed]
- Carr JL, Agnihotri S, Keightley M. Sensory processing and adaptive behavior deficits of children across the fetal alcohol spectrum disorder continuum. Alcohol Clin Exp Res. 2010;34(6):1022-32. [CrossRef] [Google scholar]
- Thomas SE, Kelly SJ, Mattson SN, Riley EP. Comparison of social abilities of children with fetal alcohol syndrome to those of children with similar IQ scores and normal controls. Alcohol Clin Exp Res. 1998;22(2):528-33. [CrossRef] [Google scholar]
- Ermer J, Dunn W. The sensory profile: a discriminant analysis of children with and without disabilities. Am J Occup Ther. 1998;52(4):283-90. [CrossRef] [Google scholar] [PubMed]
- Dunn W, Bennett D. Patterns of sensory processing in children with attention deficit hyperactivity disorder. Occup Ther J Res. 2002;22:4-15. [CrossRef] [Google scholar]
- Kientz MA, DunnW. A comparison of the performance of children with and without autism on the sensory profile. Am J Occup Ther. 1997;51:530-7. [CrossRef] [Google scholar] [PubMed]
- Rogers SJ, Hepburn S, Wehner E. Parent reports of sensory symptoms in toddlers with autism and those with other developmental disorders. J Autism Dev Disord. 2003;33:631-42. [CrossRef] [Google scholar] [PubMed]
- Baker AE, Lane A, Angley MT, Young RL. The relationship between sensory processing patterns and behavioural responsiveness in autistic disorder: a pilot study. J Autism Dev Disord. 2008;38:867–75. [CrossRef] [Google scholar] [PubMed]
- Dunn W, Myles BS, Orr S. Sensory processing issues associated with Asperger syndrome: a preliminary investigation. Am J Occup Ther. 2002;56:97-102. [CrossRef] [Google scholar] [PubMed]
- Baranek GT, Chin YH, Hess LMG, Yankee JG, Hatton DD, Hooper SR. Sensory processing correlates of occupational performance in children with fragile X syndrome: preliminary findings. Am J Occup Ther. 2002;56:538–46. [CrossRef] [Google scholar] [PubMed]
- Jones KL, Smith DW, Ullelab CN, Streissguth AP. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet. 1973;301(7815):1267-71. [CrossRef] [Google scholar] [PubMed]
- Olegård R, Sabel KG, Aronsson M, Sandin B, Johansson PR, Carlsson C, et al. Effects on the child of alcohol abuse during pregnancy. Acta Paediatr Scand Suppl. 1979;275:112-21. [CrossRef] [Google scholar] [PubMed]
- Kyllerman M, Aronson M, Sabel KG, Karlberg E, Sandin B, Olegård R. Children of alcoholic mothers. Acta Pediatr Scand. 1985;74:20-6. [CrossRef] [Google scholar] [PubMed]
- Adnams CM, Kodituwakku PW, Hay A, Molteno CD, Viljoen D, May PA. Patterns of Cognitive-Motor Development in Children With Fetal Alcohol Syndrome From a Community in South Africa. Alcohol Clin Exp Res. 2001;25:557–62. [CrossRef] [Google scholar]
- Bay B, Kesmodel U. Prenatal alcohol exposure- a systemic review of the effects on child motor function. Acta Obstet Gynecol Scand. 2011;90(3):210-26. [Google scholar]
- Kodituwakku PW, Handmaker NS, Cutler SK, Weathersby EK, Handmaker SD. Specific impairments in self-regulation in children exposed to alcohol prenatally. Alcohol Clin Exp Res. 1995;19(6):1558-64. [CrossRef] [Google scholar] [PubMed]
- Mattson SN, Goodman AM, Caine C, Delis DC, Riley EP. Executive functioning in children with heavy prenatal alcohol exposure. Alcohol Clin Exp Res. 1999;23(11):1808-15. [CrossRef] [Google scholar]
- Moghaddam B, Homayoun H. Divergent plasticity of prefrontal cortex networks. Neuropsychopharmacology. 2008;33:42-55. [CrossRef] [Google scholar] [PubMed]
- Abernathy K, Chandler LJ, Woodward JJ. Alcohol and the prefrontal cortex. Int Rev Neurobiol. 2010;91:289-319. [CrossRef] [Google scholar] [PubMed]
- Belknap JK, Crabbe JC, Young ER. Voluntary consumption of ethanol in 15 inbred mouse strains. Psychopharmacology. 1993;112:503-10. [CrossRef] [Google scholar] [PubMed]
- Whatley VJ, Johnson TE, Erwin V. Identification and confirmation of quantitative trait loci regulating alcohol consumption in congenic strains of mice. Alcohol Clin Exp Res. 1999;23(7):1262-71. [CrossRef] [Google scholar] [PubMed]
- Erecinska M, Cherian S, Silver IA. Energy metabolism in mammalian brain during development. Prog Neurobiol. 2004;73:397-445. [CrossRef] [Google scholar] [PubMed]
- Dobbing J, Sands J. Quantitative growth and development of human brain. Arch Dis Child. 1973;48:757-67. [CrossRef] [Google scholar] [PubMed]
- Sidman RL, Rakic P. Neuronal migration, with special reference to developing human brain: a review. Brain Res. 1973;62:1-35. [CrossRef] [Google scholar] [PubMed]
- Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neurosci. 2001;105(1):7-17. [CrossRef] [Google scholar] [PubMed]
- Patten AR, Fontaine CJ, Christie BR. A comparison of the different animal models of fetal alcohol spectrum disorders and their use in studying complex behaviors. Front Pediatr. 2014;2:1-19. [CrossRef] [Google scholar] [PubMed]
- Lieber C, DeCarli LM. Liquid diet technique of ethanol administration: 1989 update. Alcohol Alcohol. 1989;24(3):197-211. [Google scholar]
- Allan AM, Chynoweth J, Tyler LA, Caldwell KK. A mouse model of prenatal ethanol exposure using a voluntary drinking paradigm. Alcohol Clin Exp Res. 2003;27:2009–16. [CrossRef] [Google scholar] [PubMed]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53-63. [CrossRef] [Google scholar] [PubMed]
- Brady ML, Allan AM, Caldwell KK. A limited access mouse model of prenatal alcohol exposure that produces long-lasting deficits in hippocampal-dependent learning and memory. Alcohol Clin Exp Res. 2012;36:457–66. [CrossRef] [Google scholar] [PubMed]
- Thiele TE, Crabbe JC, Boehm II SL. “Drinking in the Dark” (DID): a simple mouse model of binge-like alcohol intake. Curr Protoc Neurosci. 2015;68:1-17. [Google scholar]
- Skorput AGJ, Gupta VP, Yeh PW, Yeh HH. Persistent interneuronopathy in the prefrontal cortex of young adult offspring exposed to ethanol in utero. J Neurosci. 2015;35(31):10977-88. [CrossRef] [Google scholar] [PubMed]
- Crabbe JC, Harris RA, Koob GF. Preclinical studies of alcohol binge drinking. Ann NY Acad Sci. 2011;1216:24-40. [CrossRef] [Google scholar] [PubMed]
- Thiele TE, Navarro M. “Drinking in the Dark” (DID) procedures: a model of binge-like ethanol drinking in non-dependent mice. Alcohol. 2014;48(3):235-41. [CrossRef] [Google scholar] [PubMed]
- Ethen MK, Ramadhani TA, Scheurle AE, Canfield MA, Wyszynski DF, Druschel CM, et al. Alcohol consumption by women before and during pregnancy. Matern Child Health J. 2009;13:274-85. [CrossRef] [Google scholar] [PubMed]
- Nadarajah B, Parnavelas JG. Modes of neuronal migration in the developing cerebral cortex. Nature Rev Neurosci. 2002;3:423-32. [CrossRef] [Google scholar] [PubMed]
- Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27(7):392-9. [CrossRef] [Google scholar] [PubMed]
- Marín O. Cellular and molecular mechanisms controlling the migration of neocortical interneurons. Eur J Neurosci. 2013;38:2019-29. [CrossRef] [Google scholar] [PubMed]
- Kroener S, Mulholland PJ, New NN, Gass JT, Becker HC, Chandler LJ. Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PLoS one. 2012;7(5):1-12. [CrossRef] [Google scholar] [PubMed]
- Morton RA, Valenzuela CF. Third trimester equivalent alcohol exposure reduces modulation of glutamatergic synaptic transmission by 5-HT1A receptors in the rat hippocampal CA3 region. Front Neurosci. 2016;10:1-7. [CrossRef] [Google scholar] [PubMed]
- DeFelipe J, Fariñas I. The pyramidal neuron of the cerebral cortex: morphological and chemical characteristics of the synaptic inputs. Prog Neurobiol. 1992;39:563-607. [CrossRef] [Google scholar] [PubMed]
- Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nature Neurosci. 2004;7(2):136-44. [CrossRef] [Google scholar] [PubMed]
- Englund C, Fink A, Lau C, Pham D, Dazza RAM, Bulfone A, et al. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25(1):247-51. [CrossRef] [Google scholar] [PubMed]
- Rakic P. Guidance of neurons migrating to the fetal monkey cortex. Brain Res. 1971;33:471-6. [CrossRef] [Google scholar] [PubMed]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong ROL, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nature Neurosci. 2001;4(2):143-50. [CrossRef] [Google scholar] [PubMed]
- Marín O, Rubenstein JLR. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441-83. [CrossRef] [Google scholar] [PubMed]
- Marin-Padilla, M. Dual origin of the mammalian neocortex and evolution of the cortical plate. Anat Embryol. 1978;152:109-26. [CrossRef] [Google scholar] [PubMed]
- Angevine JB, Sidman RL. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature. 1961;192:766-8. [CrossRef] [Google scholar] [PubMed]
- Gao P, Sultan KT, Zhang X, Shi S. Lineage-dependent circuit assembly in the neocortex. Development. 2013;140:2645-55. [CrossRef] [Google scholar] [PubMed]
- Miller MW. Effect of prenatal exposure to ethanol on the development of cerebral cortex: I. neuronal generation. Alcohol Clin Exp Res. 1988;12(3):440-9. [CrossRef] [Google scholar] [PubMed]
- Miller MW. Effect of alcohol on the generation and migration of cerebral cortical neurons. Science. 1986;233:1308-10. [CrossRef] [Google scholar] [PubMed]
- Miller MW. Effect of prenatal exposure to ethanol on neocortical development: II. Cell proliferation in the ventricular and subventricular zones of the rat. J Comp Neurol. 1989;287:326-38. [CrossRef] [Google scholar] [PubMed]
- Anderson SA, Marín O, Horn C, Jennings K, Rubenstein JLR. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128:353-6. [Google scholar]
- Parnavelas JG. The origin and migration of cortical interneurons: new vistas. Trends Neurosci. 2000;23:127-31. [CrossRef] [Google scholar] [PubMed]
- Marín O, Rubenstein JLR. A long, remarkable journey: tangential migration in the telencephalon. Nature Rev Neurosci. 2001;2:780-90. [CrossRef] [Google scholar] [PubMed]
- Cuzon VC, Yeh PW, Cheng Q, Yeh HH. Ambient GABA promotes cortical entry of tangentially migrating cells derived from the medial ganglionic eminence. Cereb cortex. 2006;16:1377-88. [CrossRef] [Google scholar] [PubMed]
- Cuzon VC, Yeh PWL, Yanagawa Y, Obata K, Yeh HH. Ethanol consumption during early pregnancy alters the disposition of tangentially migrating GABAergic interneurons in the fetal cortex. J Neurosci. 2008;28(8):1854-64. [CrossRef] [Google scholar] [PubMed]
- Skorput AGJ, Yeh HH. Chronic gestational exposure to ethanol leads to enduring aberrances in cortical form and function in the medial prefrontal cortex. Alcohol Clin Exp Res. 2016;40:1479-88. [CrossRef] [Google scholar] [PubMed]
- Beckmann H. Developmental malformations in cerebral structures of schizophrenic patients. Eur Arch Psychiatry Clin Neurosci. 1999;249:44-7. [CrossRef] [Google scholar] [PubMed]
- Akbarian S, Bunney Jr. WE, Potkin SG, Wigal SB, Hagman JO, Sandman CA et al. Altered distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase cells in frontal lobe of schizophrenics implies disturbances of cortical development. Arch Gen Psychiatry. 1993;50:169-77. [CrossRef] [Google scholar] [PubMed]
- Kato M, Dobyns W. X-linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: proposal for a new term, “interneuronopathy”. J Child Neurol. 2005;20:392-7. [CrossRef] [Google scholar] [PubMed]
- Wegiel J, Kuchna I, Nowicki K, Imaki H, Wegiel J, Marchi E, et al. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010;119:755-70. [CrossRef] [Google scholar] [PubMed]
- Simms ML, Kemper TL, Timbie CM, Bauman ML, Blatt GJ. The anterior cingulate cortex in autism: heterogeneity of qualitative and quantitative cytoarchitecture features suggests possible subgroups. Acta Neuropathol. 2009;118:673-84. [CrossRef] [Google scholar] [PubMed]
- Gleeson JG, Walsh CA. Neuronal migration disorders: from genetic diseases to developmental mechanisms. Trends Neurosci. 2000;23(8):352-9. [CrossRef] [Google scholar] [PubMed]
- Ross ME, Walsh CA. Human brain malformations and their lessons for neuronal migration. Annu Rev Neurosci. 2001;24(1):1041-70. [CrossRef] [Google scholar] [PubMed]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92(1):63-72. [CrossRef] [Google scholar] [PubMed]
- de Rouvroit CL, Goffinet AM. Neuronal migration. Mech Dev. 2001;105(1):47-56. [CrossRef] [Google scholar] [PubMed]
- Center for Disease Control and Prevention. Fetal Alcohol Spectrum Disorders (FASDs): Treatments. Atlanta (GA): Centers for Disease Control and Prevention, National Center on Birth Defects and Developmental Disabilities, Division of Birth Defects and Developmental Disabilities. Available from: https://www.cdc.gov/ncbddd/fasd/treatments.html (Accessed on February 1, 2017). [PubMed]
- Obata K, Oide M, Tanaka H. Excitatory and inhibitory actions of GABA and glycine on embryonic chick spinal neurons in culture. Brain Res. 1978;144:179-84. [CrossRef] [Google scholar] [PubMed]
- Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nature Rev Neurosci. 2002;3:728-39. [CrossRef] [Google scholar] [PubMed]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820-38. [CrossRef] [Google scholar] [PubMed]
- Delpire E. Cation-chloride cotransporters in neuronal communication. News Physiol Sci. 2000;15:309-12. [Google scholar]
- Zeisel SH, Blusztajn JK. Choline and human nutrition. Annu Rev Nutr. 1994;14:269-96. [CrossRef] [Google scholar] [PubMed]
- Thomas JD, Abou EJ, Dominguez HD. Prenatal choline supplementation mitigates the adverse effects of prenatal alcohol exposure on development in rats. Neurotoxicol Teratol. 2009;31(5):303-11. [CrossRef] [Google scholar] [PubMed]
- Thomas JD, Idrus NM, Monk BR, Dominguez HD. Prenatal choline supplementation mitigates behavioral alterations associated with prenatal alcohol exposure in rats. Birth Defects Res A Clin Mol Teratol. 2010;88(10):827-37. [CrossRef] [Google scholar] [PubMed]
- Thomas JD, Garrison M, O’Neill TM. Perinatal choline supplementation attenuates behavioral alterations associated with neonatal alcohol exposure in rats. Neurotoxicol Teratol. 2004;26:35-45. [CrossRef] [Google scholar] [PubMed]
- Ryan SH, Williams JK, Thomas JD. Choline supplementation attenuates learning deficits associated with neonatal alcohol exposure in the rat: effects of varying the timing of choline administration. Brain Res. 2008;1237:91-100. [CrossRef] [Google scholar] [PubMed]
- Monk BR, Leslie FM, Thomas JD. The effects of perinatal choline supplementation on hippocampal cholinergic development in rats exposed to alcohol during the brain growth spurt. Hippocampus. 2012;22(8):1750-7. [CrossRef] [Google scholar] [PubMed]
- Nguyen TT, Risbud RD, Mattson SN, Chambers CD, Thomas JD. Randomized, double-blind, placebo-controlled clinical trial of choline supplementation in school-aged children with fetal alcohol spectrum disorders. Am J Clin Nutr. 2016;104(6):1683-92. [CrossRef] [Google scholar] [PubMed]
- Houseknecht KL, Cole BM, Steele PJ. Peroxisome proliferator-activated receptor gamma (PPARγ) and its ligands: a review. Domest Anim Endocrinol. 2002;22:1-23 [CrossRef] [Google scholar] [PubMed]
- Kane CJM, Phelan KD, Han L, Smith RR, Xie J, Douglas JC, et al. Protection of neurons and microglia against ethanol in a mouse model of fetal alcohol spectrum disorders by peroxisome proliferator activated receptor-γ agonists. Brain Behav Immun. 2011;25(Suppl 1):S137-S145 [CrossRef] [Google scholar] [PubMed]
- Drew PD, Johnson JW, Douglas JC, Phelan KD, Kane CJM. Pioglitazone blocks ethanol induction of microglial activation and immune responses in the hippocampus, cerebellum, and cerebral cortex in a mouse model of fetal alcohol spectrum disorders. Alcohol Clin Exp Res. 2015;39(3):445-54. [CrossRef] [Google scholar] [PubMed]

