

Increased Recurrence Risk in Phelan-McDermid (22q13.3 Deletion) Syndrome: the Importance of FISH Demonstrated by a Case Series of Five Families
Renée J. Zwanenburg 1, † ![]() , Trijnie Dijkhuizen 1
, Trijnie Dijkhuizen 1![]() , Martijn J. de Groot 1
, Martijn J. de Groot 1![]() , Sheela Nampoothiri 2
, Sheela Nampoothiri 2![]() , Marjolein H. Willemsen 3
, Marjolein H. Willemsen 3![]() , Eelco Dulfer 1
, Eelco Dulfer 1![]() , Madhavan V. Thampi 4
, Madhavan V. Thampi 4![]() , Nicole de Leeuw 3
, Nicole de Leeuw 3![]() , Conny M. A. van Ravenswaaij-Arts 1, *
, Conny M. A. van Ravenswaaij-Arts 1, *![]()
- University of Groningen, University Medical Centre Groningen, Department of Genetics, Groningen, the Netherlands
- Amrita Institute of Medical Sciences and Research Centre, Department of Pediatric Genetics, Kerala, India
- Radboud University Medical Center, Department of Human Genetics, Nijmegen, the Netherlands
- Amrita Institute of Medical Sciences and Research Centre, Department of Human Cytogenetics, Kerala, India
† This author deceased in May 2018, but she collected all families and prepared the manuscript. We would like to dedicate this paper to her.
* Correspondence: Conny M. A. van Ravenswaaij-Arts ![]()
Received: July 13, 2018 | Accepted: November 15, 2018 | Published: November 22, 2018
OBM Genetics 2018, Volume 2, Issue 4 doi: 10.21926/obm.genet.1804050
Academic Editor: Thomas Liehr
Special Issue: Applications of Fluorescence in Situ Hybridization
Recommended citation: Zwanenburg RJ, Dijkhuizen T, de Groot MJ, Nampoothiri S, Willemsen MH, Dulfer E, Thampi MV, de Leeuw N, van Ravenswaaij-Arts CMA. Increased Recurrence Risk in Phelan-McDermid (22q13.3 Deletion) Syndrome: the Importance of FISH Demonstrated by a Case Series of Five Families. OBM Genetics 2018;2(4):050; doi:10.21926/obm.genet.1804050.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Background: Phelan-McDermid syndrome (PMS), or 22q13.3 deletion syndrome, is a neurodevelopmental disorder with an estimated prevalence of 1 in 10,000 to 1 in 20,000 newborns. Although it usually occurs de novo with a low recurrence risk, an increased recurrence risk is observed in some families. In this paper, we provide an overview of the underlying causes of increased recurrence risk in families with PMS and present a workflow aimed at identifying an increased recurrence risk. Methods: First, we report clinical and (cyto) genetic data for five families with an increased recurrence risk for PMS from our clinical practices. Second, we provide an overview of cytogenetically investigated Dutch families and literature cases with an increased recurrence risk. Finally, we outline which cytogenetic tests should be performed after diagnosing PMS in a proband. Results: Using fluorescent in situ hybridization (FISH), we found a parental balanced translocation in two of our five families, maternal mosaicism for a ring chromosome 22 in two families, and maternal mosaicism for a pure terminal 22q13.3 deletion in one family. In total, seven of 34 (21%) cytogenetically investigated Dutch families appeared to have an increased recurrence risk. In the medical literature, we found 28 additional families with a parental balanced translocation, and one additional family with parental mosaicism for a ring chromosome 22. Conclusions: An increased recurrence risk in PMS may be more common than currently understood, emphasizing the importance of follow-up FISH testing after PMS diagnosis in a proband. However, as low-grade parental mosaicism may still be missed, prenatal testing should always be offered.
Graphical abstract
Keywords
Phelan-McDermid syndrome; 22q13 deletion; neurodevelopment; recurrence risk; cytogenetic diagnostics; FISH
1. Introduction
Phelan-McDermid syndrome (PMS, MIM# 606232), also called 22q13.3 deletion syndrome, is the second most common subtelomeric imbalance disorder found in individuals with intellectual disability [1]. More than 1,200 cases have been reported worldwide, and the prevalence has been estimated to be between 1 in 10,000 to 1 in 20,000 newborns according to the Phelan-McDermid Syndrome Foundation [2]. Currently, there are at least 60 children and 35 adults diagnosed with a 22q13 deletion in the Netherlands. PMS is a disorder with significant impact on patients and their families. Patients present with developmental delay, particularly of language, later evolving as moderate to severe intellectual disability [3,4]. They also have problems in adaptive behaviour and exhibit features from the autism spectrum. Some patients have additional medical problems like hypotonia, feeding difficulties, seizures, and renal abnormalities [5].
PMS is caused by a deletion of the most distal part of the long arm of chromosome 22, band q13. The deletion can be the result of a pure terminal deletion, an unbalanced translocation, or can be secondary to the formation of a ring chromosome [6]. Haploinsufficiency of the SHANK3 gene located in 22q13.3 is considered the most significant (but not exclusive) contributor to the phenotype [7]. A partly overlapping phenotype has been described for interstitial deletions without the SHANK3 gene, and genes other than SHANK3 are likely to contribute to the PMS phenotype [8]. In this paper, we refer to the PMS that exhibits the phenotype associated with a terminal 22q13.3 deletion that eliminates SHANK3.
In addition to making the diagnosis and counselling the parents about the clinical features, it is important for clinicians to estimate the recurrence risk as accurately as possible and to offer prenatal diagnostics to the parents. In most patients, the terminal 22q13.3 deletion, whether pure or resulting from an unbalanced translocation, seems to have occurred de novo; therefore, the recurrence risk is theoretically almost zero [5]. However, there are underlying mechanisms that can result in a high recurrence risk for the parents of a patient with PMS. The terminal deletion could be inherited from an affected parent, although this is very rare and has been described only once in literature [9]. More likely, the deletion could be passed on by a parent who is carrier of a balanced chromosome rearrangement. Examples of this situation are an insertional translocation [10], a pericentric inversion [11], or more commonly, a balanced reciprocal translocation [7,12,13,14]. Finally, an increased recurrence risk is expected when one of the parents is mosaic for a 22q13.3 deletion (either for a pure terminal 22q13.3 deletion or for a ring chromosome 22).
Several (cyto) genetic techniques are available to diagnose a 22q13.3 deletion and to investigate whether there is an increased recurrence risk. For the latter, a combination of techniques is required (Table 1). An increased recurrence risk, however, can never be completely excluded. For example, two families have been reported in which recurrence of the 22q13.3 deletion syndrome occurred, even though FISH analysis on parental blood lymphocytes showed normal results in one family [15], as did chromosomal analysis on parental lymphocytes and buccal cells in the other family [16].
In this paper, we first reported clinical and (cyto) genetic data for five families with an increased PMS recurrence risk from our practices. Next, we provided an overview of cytogenetically investigated Dutch families and of literature cases with an increased recurrence risk. Finally, we outlined a genetic work-up to identify an increased PMS recurrence risk after diagnosing a 22q13.3 deletion in a proband.
Table 1 Techniques to detect and evaluate a 22q13 deletion.
|
Technique |
Detects |
Does not detect |
|
Conventional karyotyping, microscopic
|
Large terminal deletions and duplications Large translocations Ring chromosomes |
Small deletions, duplications and translocations Exact deletion size Parental origin of the aberrant chromosome |
|
Subtelomeric Multiplex Ligation-dependent Probe Amplification (MLPA) |
Specific subtelomeric deletions or duplications (targeted) Large 22qter imbalances can also be detected, but not sized |
Interstitial aberrations Underlying chromosome rearrangement Exact deletion size Parental origin of the aberrant chromosome |
|
Chromosome-22-specific MLPA
|
Specific deletions and duplications (targeted, depends on available markers) Large 22qter imbalances can also be detected, but not sized |
Aberrations located elsewhere Underlying chromosome rearrangement Exact deletion size Parental origin of the aberrant chromosome |
|
Metaphase Fluorescent In Situ Hybridization (FISH) |
Specific deletions or duplications (targeted) Translocations Ring chromosomes |
Non-targeted aberrations Exact deletion size Parental origin of the aberrant chromosome |
|
Array Comparative genomic hybridization (array CGH) |
Most microdeletions and duplications (genome wide, depends on the resolution) |
Underlying chromosome rearrangement Parental origin of the aberrant chromosome |
|
Single Nucleotide Polymorphism (SNP)-based array |
Most microdeletions and duplications (genome-wide, higher resolution, above detection threshold) Parental origin of the aberrant Chromosome (if parental DNA is available) |
Underlying chromosome rearrangement |
|
Whole exome sequencing (WES) |
Most microdeletions and duplications (genome-wide, high resolution, depends on algorithm) Parental origin of the aberrant chromosome (if parental DNA is available) |
Underlying chromosome rearrangement |
|
Whole genome sequencing (WGS)
|
In theory, all genomic rearrangements, but regions rich in repeats are difficult to sequence*: Microdeletions and duplications Ring chromosomes Parental origin of the aberrant chromosome (if parental DNA is available) |
|
2. Patients and Methods
Clinical and molecular data were collected from families in our centres with two affected children or with a confirmed parental mosaicism. Family 1 was evaluated at the Amrita Institute of Medical Sciences and Research Centre in Kerala (India); families 2, 3, and 5 at the University Medical Centre Groningen (the Netherlands); and family 4 at the Radboud University Medical Centre in Nijmegen (the Netherlands), with additional studies performed at the University Medical Centre Groningen.
To investigate the occurrence of increased recurrence risk in Dutch families with a 22q13.3 deletion, available cytogenetic data were collected from families either diagnosed at the Department of Genetics (clinical genetics section) of the University Medical Centre Groningen or referred to this centre from one of the other University Medical Centres in the Netherlands.
An extensive PubMed literature search was conducted (up to November 1, 2018) to find additional PMS families with an increased recurrence risk. The following terms were used: 22q13 deletion, Phelan-McDermid, and PMS. All available articles including clinical and (cyto) genetic information of patients with PMS were screened for parental translocations or mosaicism and references were checked for additional cases. Patients who were reported twice were included only once.
3. Results
3.1 Families with Increased Recurrence Risk from Our Practices
For clarity, we briefly describe the identification of the 22q13 deletion in the proband and the detection of a high recurrence risk in these families. A more detailed clinical and (cyto)genetic description of these families can be found in the Supplementary materials (Supplement 1). All genetic analyses have been performed on peripheral lymphocytes unless otherwise indicated.
Family 1. A boy, aged five years and nine months, the first child of two healthy non-consanguineous parents, was referred for evaluation because of global developmental delay and dysmorphic features. He was found to have a 46,XY karyotype, while multiplex ligation-dependent probe amplification (MLPA) analysis for common microdeletions showed a 22q13.3 deletion that was confirmed by fluorescent in situ hybridization (FISH) analysis (Supplement 1). The boy’s sister presented with developmental delay at the age of seven months. She was selectively tested for the 22q13.3 deletion using FISH, which showed that she had this familial deletion. Karyotyping of both parents revealed a balanced reciprocal translocation between chromosomes 22q13.3 and 13q32.3 in the father, and this was confirmed by FISH. Subsequent SNP array analysis showed a 22q13.32q13.33 deletion of 2.61 Mb and a 13q32.3q34 duplication of 14.06 Mb that had not been detected at karyotyping.
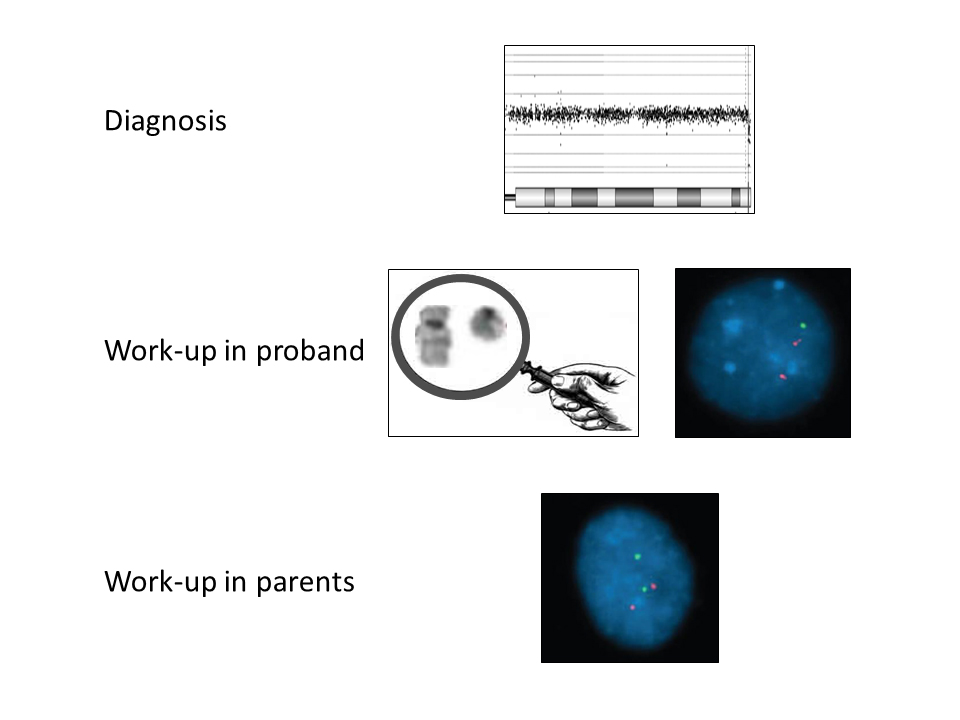
Family 2. A boy, aged three years and nine months, was referred for evaluation because of developmental delay, behavioural problems, and feeding problems. His karyotype showed no visible abnormalities; an additional array comparative genomic hybridization (array CGH) showed a terminal 22q13.33 deletion of 0.28 Mb (Supplement 1). Array CGHs of both parents were normal. Because the parents wanted to have another child, FISH analysis was performed and detected a balanced translocation between chromosomes 7p22.3 and 22q13.33 in the father. At that time, the younger brother of the proband, aged two years and four months, showed mild language and behavioural problems. He was tested by FISH and array CGH, which revealed a duplication of 22q13.33 based on a derivative chromosome 7 with an extra FISH signal for the 22q13.3 region and loss of a signal for 7p22.3. Thus, the karyotypes in the proband and his younger brother were 46,XY,der(22)t(7;22)(p22.3;q13.33)pat and 46,XY,der(7)t(7;22)(p22.3;q13.33)pat, respectively. A re-examination of the proband’s array CGH result revealed a small terminal 7p22.3 duplication of 0.17 Mb that was under the detection limit set for the software, but would have been suggestive for a translocation, if detected (Figure 1).
Figure 1 Array-CGH result on DNA from peripheral blood from the proband of family 2. A. Deletion on chromosome 22q13.33. B. Duplication on chromosome 7p22.3.
Family 3. A boy, aged three years and three months, was diagnosed with PMS by his paediatrician after being referred because of a history of global developmental delay and behavioural problems. Array CGH analysis showed a terminal 22q13.32q13.33 deletion of 2.03 Mb in the boy, but not in his parents (Supplement 1). Later, his younger sister was also tested, because of parental concerns about her behavioral development. Array-analysis showed that she also had the 22q13.32q13.33 deletion. Metaphase FISH analysis in both children showed that this 22q13.3 deletion was a pure terminal deletion (i.e. not associated with a ring chromosome 22). Subsequent FISH analysis on lymphocytes in both parents showed normal results. The parental origin of the deleted chromosome 22 was determined by SNP array analysis of patients and parents, and the maternal 22q13.32q13.33 allele appeared to be deleted in both siblings. Subsequent FISH investigations on parental buccal and urinary cells showed a mosaic 22q13.3 deletion in 20% of the mother’s urinary cells, thus confirming maternal mosaicism (of note, this maternal mosaicism was not detected in buccal cells).
Family 4. A girl, the second child of healthy parents, was seen at the neonatal intensive care unit at the age of six weeks because of intrauterine growth restriction and several dysmorphic features, following the mother's pregnancy that was complicated by maternal hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome. Both the girl and her parents were tested by SNP array analysis, which revealed a de novo terminal 22q13.2q13.33 deletion of 9.23 Mb on the maternal allele (Supplement 1). Karyotyping showed that this 22q13.2q13.33 deletion resulted from a ring chromosome 22: 46,XX,r(22)(p11q13.2). The first child, a boy, was born prematurely and had died 18 hours postnatally of severe infantile respiratory distress syndrome. Post-mortem SNP array analysis on stored fibroblasts of the boy revealed the same 22q13.2q13.33 deletion on the maternal allele. Subsequent FISH analysis on maternal buccal cells showed mosaicism for the 22q13 deletion in approximately 5% (5/107) of the investigated cells. Of note, this deletion could not be detected in maternal lymphocytes using FISH analysis, nor in maternal urinary cells using SNP array analysis.
Family 5. A boy, aged two years and three months, was referred by his paediatrician for analysis of his developmental delay and several mild dysmorphic features. The boy’s older brother was healthy and had normal development. Conventional karyotyping in the younger brother showed a ring chromosome 22. Additional array CGH revealed a terminal 22q13.31q13.33 deletion of 2.98 Mb. Furthermore, the array analysis showed an interstitial 15q11.2 deletion of 1.59 Mb (Supplement 1). Parental FISH-analysis revealed that the boy’s healthy mother was mosaic for the ring chromosome 22 (with a low-grade mosaicism of 3% in maternal blood lymphocytes).
3.2 Families with PMS in the Netherlands
In total, we were able to collect clinical and cytogenetic information of 46 children with PMS from 45 families diagnosed in one of the Dutch University Medical Centres (including four of the five families reported above). We retrieved parental cytogenetic data from 34 of these families. In three of the 34 (9%) families, a parental balanced translocation was found (family 2 in this paper, one previously published family [4], and one unpublished family). In four of the 34 (12%) families, parental mosaicism for a pure terminal 22q13.3 deletion or for a ring chromosome 22 was found (families 3, 4, and 5 of this report, and one previously published family [16]). Thus, an increased recurrence risk was observed in seven of 34 (21%) Dutch PMS families for which parental cytogenetic testing data was available.
3.3 Literature Cases with an Increased PMS Recurrence Risk
We found 36 articles reporting 66 cases with a 22q13.3 deletion possibly associated with an increased recurrence risk, i.e., 46 patients with translocations involving 22q13.3 and 20 patients with mosaicism (for either a pure terminal 22q13.3 deletion or a ring chromosome 22). From the 46 translocation patients, we excluded two patients with insufficient information, three patients with a more complex translocation, and 13 patients with a de novo translocation. Results of the remaining 28 patients with a parental translocation are summarized in Table 2. (For de novo translocations and their references, see Supplement 2). Of these 28 inherited translocations, thirteen were maternal, seven were paternal, and for the remaining eight, the sex of the carrier parent remained unclear [6,7,12,13,14,17,18,19,20,21,22,23,24,25,26]. Chromosomes involved in the inherited translocations were 1 (n=2), 3 (n=9), 8 (n=1), 10 (n=2), 11 (n=2), 12 (n=3), 13 (n=1), 14 (n=2), 16 (n=1), 17 (n=2), 19 (n=2), and 20 (n=1).
Table 2 Familial translocations and mosaicism reported in literature.
|
Reference |
Origin |
Karyotype parent |
|
Tachdjian 1992 |
mat |
t(11;22)(q23.3;q13.2) |
|
Smith 1996 |
mat |
t(17;22)(p13.3;q13.3) |
|
Doheny 1997 |
mat |
t(1;22)(q44;q13.32), case 1 |
|
Praphanphoj 2000 |
mat |
t(19;22)(q13.42;q13.31) |
|
M. Phelan 2001 |
mat |
t(1;22)(q44;q13.32) |
|
|
mat |
t(14;22)(q32.31;q13.33) |
|
|
mat |
t(19;22)(q13.42;q13.31) |
|
|
pat |
t(20;22)(p13.2;q13.3) |
|
Rodriguez 2003 |
pat |
t(12;22)(q24.31;q13.3), case 1 |
|
|
mat |
t(12;22)(q24.3;q13.3), case 2 + 3 |
|
Luciani 2003 |
pat |
t(14;22)(p11;q13), case 30 |
|
Wilson 2003 |
unknown |
t(13;22)(p12;q13.3), case 57 |
|
Manning 2004 |
mat |
t(10;22)(q26.1;q13.3) |
|
|
pat |
t(17;22)(q25.3;q13.3) |
|
Chen 2005 |
pat |
t(16;22)(q12.1;q13.3) |
|
Toruner 2009 |
pat |
t(8;22)(q24.3;q13.3), case 1 |
|
Soorya 2013 |
mat |
t(11;22)(q23;q11.2), case 27 |
|
Liu 2016 |
mat |
t(3;22)(q27;q13.3) |
|
Zhang 2017 |
pat and unknown |
t(3;22)(q28;q13.3), 8 patients, 6 carriers |
|
Coci 2017 |
Mat |
t(10;22)(q26.13;q13.32), patient II.2 |
|
Reference |
Origin, % mosaicism |
Karyotype parent |
|
Jobanputra 2009* |
mat, 7% |
r(22)(5) / 46,XX(65) and 46,XX,r(22)(D22S75+,BCR+,ARSA+,qter+) |
mat, maternal; pat, paternal. *karyotype of the daughter 46,XX,r(22)(p13q13.1).ish r(22)(acrop+,D22S75+,BCR+,ARSA-,qter-).
From the 20 patients with a mosaic deletion, we excluded five patients with insufficient information and 14 patients with de novo mosaicism (seven mosaic pure terminal 22q13.3 deletions and seven mosaic ring chromosomes 22, see Supplement 2). In the one remaining family, a healthy mother with mosaicism for a “balanced” ring chromosome 22 in 7% of her lymphocytes (Table 2) passed a ring chromosome 22 with a terminal 22q13.3 deletion on to her affected daughter [27]. Apart from this family, we did not find any literature cases in which a parent with mosaicism for a pure terminal 22q13.3 deletion or a ring chromosome 22 passed the chromosome abnormality on to his or her children.
4. Discussion
This paper highlights the importance of estimating the recurrence risk after diagnosing a 22q13.3 deletion in index patients. Finding a parental origin and an increased recurrence risk for PMS in five families in our clinical practice prompted us to study the recurrence risk in further Dutch PMS families. In 21% of the families for which cytogenetic data was available, an increased recurrence risk was observed. However, in two of the families from our clinical practice, the more elaborate parental studies were initiated because of the diagnosis of a second child with PMS, resulting in a selection bias. Still, excluding these two families leaves an increased recurrence risk in 15% of the reported families.
To our knowledge, the frequency of an increased recurrence risk in PMS has not yet been studied systematically. Literature reports were found of at least 29 families with an increased PMS recurrence risk due to a parental chromosome abnormality. This number may be an overrepresentation due to publication bias. On the other hand, it may also be an underestimation due to incomplete cytogenetic work-up in affected families, particularly regarding FISH analysis in different tissue types aimed at detecting low-grade mosaicism. The most commonly published cause of an increased recurrence risk for PMS is a parental balanced translocation (Table 2). We observed a parental balanced translocation in two of our five families. In one of these families, the translocation was identified by FISH and karyotyping only after recurrence of the familial 22q13.3 deletion (family 1, Supplement 1), which highlights the importance of targeted metaphase FISH analysis in parents after diagnosing a 22q13.3 deletion in a child. Notably, FISH analysis allows detection of reciprocal rearrangements below the array detection threshold (as exemplified by family 2), a translocation of 22q13.3 to the satellites of an acrocentric chromosome, or a (balanced) 22q13.3 insertion in another chromosome. If a balanced translocation is found in one of the parents, prenatal testing options should be discussed and cascade screening should be offered. Cascade screening is important, since a balanced translocation may not be recognized in families, and confers not only a high recurrence risk, but also an increased risk of recurrent miscarriages.
A second, rarely reported cause of increased recurrence risk is parental mosaicism for the underlying chromosome 22 abnormality (either a pure terminal 22q13.3 deletion or a ring chromosome 22). Tabolacci et al. and Verhoeven et al. reported a 22q13.3 deletion of maternal origin based on polymorphic marker analysis in families with two affected siblings; this indicated parental mosaicism, but the mosaicism itself could not be detected [15,16]. Jobanputra et al. reported a proband with a ring chromosome 22 and FISH-confirmed mosaicism of a “balanced” ring chromosome 22 in the proband’s mother [27]. To our knowledge, we are the first to report on cytogenetically confirmed parental mosaicism for a pure terminal 22q13.3 deletion (in one family) and cytogenetically confirmed parental mosaicism for an unbalanced ring chromosome 22 (in two families).
Based on the limited reports on parental mosaicism for an abnormal chromosome 22 as a cause of increased recurrence risk, it is easy to hypothesize that such parental mosaicism is rare. However, we noted that systematic, more extensive FISH testing (including FISH testing on non-blood cells) was often not performed in the reported literature cases. As exemplified by our cases and the available literature, low-grade parental mosaicism may be missed when only performing genetic analyses on parental lymphocytes (in particular when FISH analysis is not performed).
In two of the families from our practices (families 3 and 4), parental mosaicism was suspected based on recurrence within the same family. In both families, while SNP array analysis showed maternal origin of the abnormal chromosome 22, it was not able to detect the (presumably low-grade) mosaicism in maternal lymphocytes. Subsequent targeted FISH analysis confirmed mosaicism for the deletion in maternal urinary cells (20%) in one family and in maternal buccal cells (5%) in the other. In our third family with parental mosaicism, a ring chromosome 22 was identified in the proband and FISH analysis of maternal lymphocytes identified a low-grade mosaicism (3%) for the ring chromosome 22.
We can compare the prevalence of parental translocations (9%) and mosaicism (11%) in PMS with other terminal deletion syndromes. In 4p deletion (Wolf-Hirschhorn) syndrome, for example, 10-15% of the deletions are estimated to be the result of a parental balanced translocation [28]. However, this high number of unbalanced translocations is partly caused by one of the few recurrent translocations found in humans: a t(4p;8p) that occurs due to the presence of low copy repeats at the breakpoints that mediate non-allelic homologous recombination [29]. As far as we know, parental mosaicism for the 4p deletion (40%) has been reported only once, but this parent also had a balanced translocation in the remainder of the cells [30]. In 5p deletion (Cri-du-chat) syndrome, three out of 80 (4%) deletions were the result of a parental translocation [31], while parental mosaicism (4-7%) has been reported only once [32]. So compared to PMS, the percentage of deletions caused by a parental balanced translocation is lower in 5p deletion syndrome and slightly higher in 4p deletion syndrome, but reports of parental mosaicism in 4p and 5p deletion syndrome are rare.
Each (cyto) genetic technique has both advantages and disadvantages, and a combination of techniques is often required to identify the deletion, determine its size and gene content, unravel the underlying mechanism, and establish the recurrence risk (Table 1). To optimize the diagnostic process, we propose the following workflow (summarized in Figure 2). When a 22q13.3 deletion is first suspected (based on conventional karyotyping or first detected by MLPA analysis or FISH analysis), subsequent array analysis should be performed in the proband to further characterize the deletion. This array analysis may also be used to look for associated duplications that may suggest an underlying unbalanced translocation. If a translocation is suggested by array analysis in the proband, his or her chromosomes should be investigated by FISH to confirm the translocation and to determine which probes can be used for the detection of a balanced translocation in the parents. If an isolated deletion is found, karyotyping allows for the detection of a ring chromosome 22, which is important since the existence of a ring chromosome 22 is associated with an increased risk of developing neurofibromatosis type 2-associated tumours. This increased tumour risk results from the mitotic instability of the ring chromosome 22 (with associated loss of one copy of the NF2 gene, which is located on chromosome 22), leading to haploinsufficiency of the NF2 gene. When a somatic pathogenic mutation occurs in the remaining NF2 gene, the affected cell(s) may give rise to NF2-associated tumours. [33]. Therefore, the detection of a ring chromosome 22 has clinical consequences, as periodic MRI screening is advised.
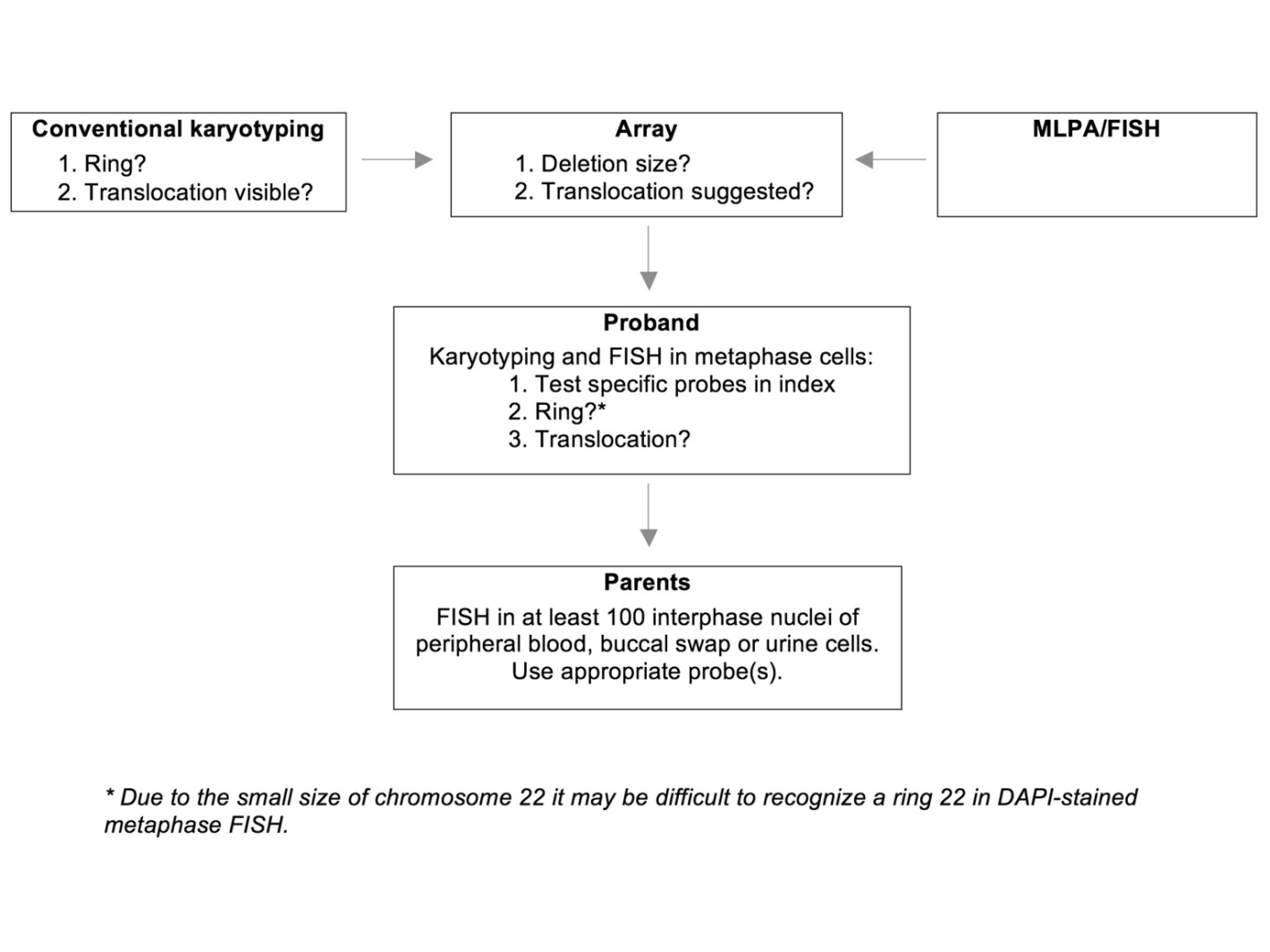
Figure 2 Workflow for the identification of an increased PMS recurrence risk. Please note that this workflow may start using either conventional karyotyping, array analysis or MLPA/FISH.
Based on our findings, we strongly advise to always perform follow-up FISH testing after diagnosing a 22q13.3 deletion. Currently, as a first step, FISH analysis on peripheral blood cells of both parents is the technique of choice. If FISH analysis on parental lymphocytes reveals mosaicism for the underlying chromosome 22 abnormality, additional FISH analyses in the parents are not required. If FISH analysis on parental lymphocytes does not show parental mosaicism, we advise to perform follow-up FISH analysis of parental buccal cells and/or urinary cells, as low-grade parental mosaicism may not be detected by FISH analysis on parental lymphocytes only. Regardless of the results of FISH analyses in the parents, prenatal screening should always be offered.
5. Conclusions
In conclusion, based on our experience and on the literature, there is evidence for an increased recurrence risk in 15 to 20% of families with Phelan-McDermid syndrome. We therefore emphasize the importance of thorough follow-up studies, including metaphase and interphase FISH in index patients and their parents. Furthermore, as low-grade parental mosaicism for an underlying chromosomal abnormality may be missed, prenatal testing should always be offered in subsequent pregnancies.
Acknowledgments
We thank all parents and children for their contribution to this study. We also thank K. Heeres (University Medical Centre Groningen, Department of Genetics) for his help with the FISH experiments of family 3, and Kate McIntyre, language editor, for revision of the manuscript.
Additional Materials
The following additional materials are uploaded at the web page of this paper.
1. Supplement 1: Clinical description and molecular and cytogenetic results of the families.
2. Supplement 2: De novo translocations and mosaicism reported in literature.
Author Contributions
Renée Zwanenburg collected all patient material, performed experiments and wrote the manuscript, Trijnie Dijkhuizen collected information from Dutch laboratories and assisted with experiments and writing the manuscript, Martijn de Groot performed the additional literature review, added patient information and assisted in the revision of the manuscript, Sheela Nampoothiri provided clinical information on patients from India and assisted with writing the manuscript, Marjolein Willemsen and Eelco Dulfer provided clinical information on Dutch patients and assisted with writing the manuscript, Madhavan Thampi performed experiments in the Indian patients, Nicole de Leeuw assisted with experiments and writing the manuscript, Conny van Ravenswaaij-Arts initiated and supervised the study and the manuscript.
Funding
This work was supported by a grant from the Netherlands Organization for Health Research and Development (ZonMw 113-20-2009 to R.Z and C.v.R.).
Competing Interests
The authors have declared that no competing interests exist.
References
- Delahaye A, Toutain A, Aboura A, Dupont C, Tabet AC, Benzacken B, et al. Chromosome 22q13.3 deletion syndrome with a de novo interstitial 22q13.3 cryptic deletion disrupting SHANK3. Eur J Med Genet. 2009; 52: 328-332. [CrossRef]
- Costales JL, Kolevzon A. Phelan-McDermid Syndrome and SHANK3: Implications for Treatment. Neurotherapeutics. 2015; 12: 620-630. [CrossRef]
- Phelan K, McDermid HE. The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol Syndromol. 2012; 2: 186-201.
- Zwanenburg RJ, Ruiter SAJ, Van den Heuvel ER, Flapper BCT, Van Ravenswaaij-Arts CMA. Developmental phenotype in Phelan-McDermid (22q13.3 deletion) syndrome: a systematic and prospective study in 34 children. J Neurodev Disord. 2016; 8: 16. [CrossRef]
- Sarasua SM, Dwivedi A, Boccuto L, Chen CF, Sharp JL, Rollins JD, et al. 22q13.2q13.32 genomic regions associated with severity of speech delay, developmental delay, and physical features in Phelan–McDermid syndrome. Genet Med. 2013; 16: 318-328. [CrossRef]
- Luciani JJ, de Mas P, Depetris D, Mignon-Ravix C, Bottani A, Prieur M, et al. Telomeric 22q13 deletions resulting from rings, simple deletions, and translocations: cytogenetic, molecular, and clinical analyses of 32 new observations. J Med Genet. 2003; 40: 690-696. [CrossRef]
- Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, et al. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003; 40: 575-584. [CrossRef]
- Disciglio V, Rizzo Lo C, Mencarelli MA, Mucciolo M, Marozza A, Di Marco C, et al. Interstitial 22q13 deletions not involving SHANK3 gene: a new contiguous gene syndrome. Am J Med Genet. 2014; 164A: 1666-1676. [CrossRef]
- Denayer A, Van Esch H, de Ravel T, Frijns JP, Van Buggenhout G, Vogels A, et al. Neuropsychopathology in 7 patients with the 22q13 deletion syndrome: Presence of bipolar disorder and progressive loss of skills. Mol Syndromol. 2012; 3: 14-20. [CrossRef]
- Slavotinek A, Maher E, Gregory P, Rowlandson P, Huson SM. The phenotypic effects of chromosome rearrangement involving bands 7q21.3 and 22q13.3. J Med Genet. 1997; 34: 857-861. [CrossRef]
- Jafri F, Fink J, Higgins RR, Tervo R. 22q13.32 deletion and duplication and inversion in the same family: a rare occurrence. ISRN Pediatrics. 2011; 829825. [CrossRef]
- Phelan MC, Rogers RC, Saul RA, Stapleton GA, Sweet K, McDermid H, et al. 22q13 deletion syndrome. Am J Med Genet. 2001; 101: 91-99. [CrossRef]
- Rodríguez L, Martinez Guardia N, Herens C, Jamar M, Verloes A, Santos Munoz J, et al. Subtle trisomy 12q24.3 and subtle monosomy 22q13.3: Three new cases and review. Am J Med Genet. 2003; 122A: 119-124. [CrossRef]
- Manning MA, Cassidy SB, Clericuzio C, Cherry AM, Schwartz S, Hudgins L, et al. Terminal 22q deletion syndrome: a newly recognized cause of speech and language disability in the autism spectrum. Pediatrics. 2004; 114: 451-457. [CrossRef]
- Tabolacci E, Zollino M, Lecce R, Sangiorgi E, Gurrieri F, Leuzzi V, et al. Two brothers with 22q13 deletion syndrome and features suggestive of the Clark-Baraitser syndrome. Clin Dysmorphol. 2005; 14: 127-132. [CrossRef]
- Verhoeven WM, Egger JI, Willemsen MH, de Leijer GJ, Kleefstra T. Phelan-McDermid syndrome in two adult brothers: atypical bipolar disorder as its psychopathological phenotype? Neuropsychiatr Dis Treat. 2012; 8: 175-179. [CrossRef]
- Tachdjian G, Muti C, Gaudelus J, Druart L, Martin B, , Tamboise E, et al. Unbalanced karyotype due to adjacent 1 segregation of t(11;22)(q23.3;q13.2). Ann Genet. 1992; 35: 231-233.
- Smith DP, Floyd M, Say B. Detection of a familial cryptic translocation by fluorescent in situ hybridisation. J Med Genet. 1996; 33: 84. [CrossRef]
- Doheny KF, McDermid HE, Harum K, Thomas GH, Raymond GV. Cryptic terminal rearrangement of chromosome 22q13.32 detected by FISH in two unrelated patients. J Med Genet. 1997; 34: 640-644. [CrossRef]
- Praphanphoj V, Goodman BK, Thomas GH, Raymond GV. Cryptic subtelomeric translocations in the 22q13 deletion syndrome. J Med Genet. 2000; 37: 58-61. [CrossRef]
- Chen C-P, Hsu C-Y, Huang J-K, Lee C-C, Chen W-L, Wang W. Prenatal diagnosis of partial trisomy 16q and distal 22q13 deletion associated with dolichocephaly and frontal bossing on second-trimester ultrasound. Prenat Diagn. 2005; 25: 964-966. [CrossRef]
- Toruner GA, Kurvathi R, Sugalski R, Shulman L, Twersky S, Pearson PG, et al. Copy number variations in three children with sudden infant death. Clin Genet. 2009; 76: 63-68. [CrossRef]
- Soorya L, Kolevzon A, Zweifach J, Lim T, Dobry Y, Schwartz L, et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol Autism. 2013; 4: 18. [CrossRef]
- Liu HY, Huang J, Li T, Wu D, Wang HD, Wang Y, et al. Clinical and molecular cytogenetic analyses of four patients with imbalanced translocations. Molec Cytogenet. 2016; 9: 31 [CrossRef]
- Zhang K, Dong R, Huang Y, Yang Y, Zhang H, Zhang Y, et al. [Clinical and molecular cytogenetic analysis of a family with mental retardation caused by an unbalanced translocation involving chromosomes 3 and 22]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2017; 34: 30-34.
- Coci EG, Auhuber A, Langenbach A, Mrasek K, Riedel J, leenen A, et al. Novel unbalanced translocation affecting the long arms of chromosomes 10 and 22 cause complex syndromes with very severe neurodevelopmental delay, speech impairment, autistic behavior, and epilepsy. Gytogenet Genome Res. 2017; 151: 171-178. [CrossRef]
- Jobanputra V, Ash E, Anyane-Yeboa K, Warburton D, Levy B. Changes in an inherited ring (22) due to meiotic recombination? Implications for genetic counseling. Am J Med Genet. 2009; 149A: 1310-1314. [CrossRef]
- Zollino M, Murdolo M, Marangi G, Pecile V, Galasso C, Mazzanti L, et al. On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C Semin Med Genet. 2008; 148C: 257-269. [CrossRef]
- South ST, Whitby H, Battaglia A, Carey JC, Brothman AR. Comprehensive analysis of Wolf-Hirschhorn syndrome using array CGH indicates a high prevalence of translocations. Eur J Hum Genet. 2008; 16: 45-52. [CrossRef]
- Fryns JP, Smeets E, Devriendt K, Petit P. Wolf-Hirschhorn syndrome with cryptic 4p16.3 deletion and balanced/unbalanced mosaicism in the mother. Ann Genet. 1998; 41: 73-76.
- Mainardi PC, Perfumo C, Calì A, Coucourde G, Pastore G, Cavani S, et al. Clinical and molecular characterisation of 80 patients with 5p deletion: genotype-phenotype correlation. J Med Genet. 2001; 38: 151-158. [CrossRef]
- Philip J, Brandt NJ, Friis-Hansen B, Mikkelsen M, Tygstrup I. A deleted B chromosome in a mosaic mother and her cri du chat progeny. J Med Genet. 1970; 7: 33-36. [CrossRef]
- Zirn B, Arning L, Bartels I, Shoukier M, Hoffjan S, Neubauer B, et al. Ring chromosome 22 and neurofibromatosis type II: proof of two-hit model for the loss of the NF2 gene in the development of meningioma. Clin Genet. 2012; 81: 82-87. [CrossRef]
