

Epigenetic Regulation by Androgen Receptor in Prostate Cancer
Ken-ichi Takayama 1, 2,*![]()
- Department of Functional Biogerontology, Tokyo Metropolitan Institute of Gerontology, Tokyo, Japan
- Department of Geriatric Medicine, the University of Tokyo, Tokyo, Japan
* Correspondence: Ken-ichi Takayama ![]()
Received: May 26, 2018 | Accepted: October 16, 2018 | Published: November 02, 2018
OBM Genetics 2018, Volume 2, Issue 4 doi: 10.21926/obm.genet.1804047
Academic Editors: Stéphane Viville and Marcel Mannens
Special Issue: Epigenetic Mechanisms in Health and Disease
Recommended citation: Takayama K. Epigenetic Regulation by Androgen Receptor in Prostate Cancer. OBM Genetics 2018;2(4):047; doi:10.21926/obm.genet.1804047.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Prostate cancer is the most common cancer among men in the world. Androgen receptor (AR), acting as a nuclear receptor, facilitates ligand-dependent transcriptional activation in the nucleus. Androgen deprivation therapy (ADT) is used for the treatment of advanced prostate cancer because androgen and AR signaling drive prostate tumor growth and anti-apoptotic function. Resistance to ADT in most tumors develops quickly; thus, AR continues to be active in relapsed tumors called castration-resistant prostate cancer (CRPC). Therefore, it is important to investigate the transcriptional mechanisms of AR and its downstream signaling. Recent studies have shown the central role of chromatin structure and histone modifications in AR-mediated gene regulation. Furthermore, AR functions through interaction with several tissue-specific transcription factors including forkhead box protein A1 (FOXA1). Interestingly, non-coding RNAs, mainly classified as long non-coding RNAs (lncRNAs) and micro RNAs (miRNAs), modulate epigenetic status to promote AR function directly or indirectly and have central roles in prostate cancer progression. This review focuses on the involvement of AR in epigenetic regulation of the development and progression of prostate cancer.
Keywords
Androgen receptor; epigenetic; non-coding RNA; prostate cancer; testosterone; transcription
1. Introduction
Prostate cancer is one of the major causes of cancer morbidity and mortality in men worldwide. Androgens promote proliferation of epithelial or cancer prostate cells [1,2]. Currently, early screening for prostate cancer is based on digital rectal examination and an analysis of the level of serum prostate-specific antigen, a protein expressed as a representative androgen-regulated gene [3]. When screening tests indicate a suspected cancer, the actual diagnosis of prostate cancer can be made with an ultrasound and biopsy [3]. Since androgen signals are necessary for the progression of both localized and metastatic prostate cancer, androgen deprivation therapy (ADT), which blocks androgen binding to androgen receptor (AR) or reduces the serum androgen level, is a successful initial strategy for treating prostate cancer at advanced stages [4]. However, relapse and the progression to castration-resistant prostate cancer (CRPC) or hormone-refractory prostate cancer are observed in most of these cancers [1,2].
Androgens (male sex hormones) exert their functions by binding to the AR, which is a member of the nuclear receptor superfamily [5]. The two most abundant androgens, testosterone and dihydrotestosterone (DHT), bind to AR and activate transcription of its target gene in the nucleus [1]. The androgen hormone is responsible for various human physiological phenomena throughout the body [6]; For example, as a male sex hormone, testosterone is required for the establishment of adult sexual function to induce the differentiation of the male reproductive system [6]. In addition, androgens have various anabolic functions in several tissues, such as bone and muscle [6]. The enzyme 5α-reductase is responsible for the conversion of testosterone to dihydrotestosterone (DHT) in cells [5]. DHT binds to AR with an even higher affinity for activation than testosterone [7]. Notably, AR has an important role in the initiation, development, and subsequent progression of prostate cancer [1,2]. This review focuses on the molecular function of AR in epigenetic processes, mainly histone modifications and non-coding RNAs, in prostate cancer pathogenesis and progression, and addresses further modalities of treatments that target epigenetic phenomena in prostate cancer.
2. Functions of AR in Prostate Cancer: Aberrant AR Activation in Advanced Prostate Cancer
AR functions as a ligand-dependent transcription factor [1,5,8]. In the absence of ligand, AR forms a complex with molecular chaperones and co-chaperones from the heat-shock protein family, mainly in the cytoplasm (Figure 1). Upon androgen treatment, a conformational change of the complex occurs to facilitate nuclear translocation of AR. Homodimers of AR bind to genomic regions in the nucleus that contain the specific sequence motif, androgen responsive element (ARE) [8,9], to modify the epigenetic conditions surrounding the AR-binding regions that function to activate gene expression [8,9,10]. In general, nuclear receptors possess several domains: N-terminal domain (NTD), DNA-binding domain, and ligand-binding domain (LBD) [11,12,13]. Moreover, the transcriptional activation function 1 (AF1) domain in the NTD is responsible for transcriptional activation with or without ligand binding [13,14]. The activation function 2 (AF2) domain located in the LBD is involved in the interaction between AR and co-regulators containing the LXXLL motif [12]. Interestingly, point mutations mapped to the LBD have been found and are involved with resistance to hormone treatment in prostate cancer [15,16]. In this case, the interplay between these domains exerts AR transcription and epigenetic functions.
Suppressing circulating testosterone to castration levels for the treatment of advanced prostate cancer does not decrease androgens sufficiently from the prostate tumor microenvironment [2,3,17]; therefore, AR remains active in CRPC tumors, and residual androgen levels remain within the range capable of activating AR [1,2,17,18]. Persistent AR activation provides a compelling rationale for developing more effective strategies to inhibit AR signals [18,19,20,21,22,23]. A new type of drug, called abiraterone acetate, which targets androgen synthesis and is a potent inhibitor of CYP17, blocks testosterone production from cholesterol [19]. Enzalutamide (MDV3100) is a newly developed endocrine treatment agent with anti-tumor activity [20], and it significantly prolonged the survival of men with metastatic CRPC after chemotherapy [21]. Mechanistically, it is an AR blocker that inhibits AR nuclear translocation, DNA binding, and interaction with co-activators. Although these new drugs show impressive results, a majority of CRPC cases eventually develop resistance to the drugs [22]. Another new AR blocker, apalutamide (ARN-509), is under development for the treatment of CRPC. A recent clinical trial showed that apalutamide treatment improved the metastasis-free survival frequency and inhibited symptomatic progression of disease [23], although it produced a higher than usual rate of side effects, such as fractures. Overall, these new anti-androgen drugs could be promising for the treatment of CRPC.
Overexpression of AR by hypersensitivity to androgen hormones, intratumoral steroidogenesis, gene amplification, point mutations, or production of AR variants (AR-Vs) are assumed to be the primary causes for enhanced AR signals [17,18,24,25]. The mRNA of AR is alternatively spliced to produce AR-Vs and most AR-Vs are missing the LBD. However, the retained NTD drives transcription without androgen [26,27,28]. Interestingly, AR-Vs preferentially bind to distinct genomic regions of full-length AR [26]. Global analysis of AR-V binding sites revealed that the AR-V target genes are upregulated in CRPC specimens [27]. The expression of AR-V7 is most frequently observed AR-V in CRPC tissues [27], and increased AR expression enhances the reactivity of prostate cancer cells to low androgen concentrations (castrate level) and promotes AR-targeting drug resistance [28]. To overcome this problem, new drugs have been developed that target AR NTD, such as EPI (ralaniten acetate). EPI effectively inhibits AR activity that is enhanced by overexpressed co-activators, AR mutation, and AR-V7 [29], although a recent clinical trial of EPI-506 was discontinued due to side effects [30]. Thus, more effective and safer inhibition of AR and AR-V activity may improve the clinical management of CRPC.
3. The Roles of Collaborative Transcription Factors for AR-Meditated Epigenetic Regulation
The functional genomic regions that AR binds to are dependent on chromatin accessibility as well as genome sequence [1,5,8,10]. Generally, the ChIP-sequence (ChIP-seq) method, which uses high-throughput powerful sequencers, has been utilized to determine endogenous transcription-factor binding sites in cells [31]. By investigating the functions of androgen-regulated genes located near AR binding sites (ARBSs), new prostate cancer associated factors have been identified [32,33,34,35,36,37,38,39]. Interestingly, AR ChIP-seq showed that more ARBSs were present in androgen-dependent prostate cancer cells that overexpress AR compared to parental cells, even when only treated low level androgen [40], suggesting that the higher expression of AR sensitizes the receptor recruitment to the genome.
Moreover, analysis of enriched motifs surrounding the ARBSs has identified transcription partners that interact with AR; these including the following: forkhead box protein A1 (FOXA1) [10,41], FOXP1 [39], ETS-related gene (ERG), GATA binding protein 2 (GATA2) [41], octamer-Binding Protein 1 (Oct1) [41,42], and runt Related Transcription Factor 1 (RUNX1) [43]. All of these function as important factors in the AR-driven transcriptional program. Among these factors, a chromatin-opening transcription factor called FOXA1 directly occupies the chromatin to loosen the nucleosomal complex in the region and increase accessibility for other epigenetic factors [41]. FOXA1 protein has been shown to physically associate with the AR protein and activate the AR-mediated transcription of prostate cancer associated genes [10]. In addition, ChIP-seq analysis used to identify ARBSs in CRPC tissues revealed that most of them are not present in cell lines [44]. In this study, transcription factor motifs such as E2F, Myc, and STAT were significantly enriched in these CRPC-specific ARBSs. Many adjacent genes, in vivo, were members of a restricted set of AR-regulated genes. The differing AR signals between cell lines and human tissues could be explained by genetic or epigenetic alterations of AR-associated factors (such as FOXA1 mutation or retinoblastoma protein [RB] inactivation), or by integration of paracrine [cytokine] signaling events in cancer tissues that impact other transcription factors to regulate AR activity [44]. Another study showed the colocalization of FOXA1 and homeobox B13 (HOXB13) at a set of ARBSs in prostate tumor tissues [45]. Taken together, these studies suggest that AR collaborating factors reprogrammed the chromatin state to recruit AR and its associated epigenetic factors and promoted prostate tumor progression.
4. Classification of Epigenetic Regulations
A highly ordered structure, called chromatin, consists of histones and other proteins wrapped around DNA [46]. A histone octamers (two pairs each of H2A, H2B, H3, and H4) and DNA form a unit called a nucleosome. Two major patterns of epigenetic changes, DNA methylation and histone modifications, have also been analyzed in prostate cancer cells [8]. DNA methylation, a representative epigenetic marker, is formed by the addition of a methyl group to the 5’ position of cytosine (5-mC) within the genome. DNA is methylated in a spatial and temporal context throughout the genome, particularly in enhancer/promoter regions [47], and DNA methyltransferases (DNMTs) are responsible for this process [47]. For histone modification, amino acids of N-terminal histone tails (mainly lysine, arginine, serine, and threonine residues) serve as substrates to be modified with post-translational acetylation, phosphorylation, methylation, ubiquitination, sumoylation, and deamination changes [48]. The interaction of nucleosomes with transcription factors or other proteins involved in transcription is affected by histone modification patterns in epigenetic biological events. For example, the histone H3 is one of the major histones and is highly modified epigenetically. The representative epigenetic patterns of histone H3 tails include methylation of lysine on position 9 (H3K9) and H3K27, which indicates condensed chromatin and silenced loci. In contrast, the methylation (me) of H3K4 and H3K36 is associated with activated chromatin structures. Similarly, acetylation (ac) of lysine residues of H3 is also correlated with activated promoter or enhancer regions [8,9,11]. Thus, multiple histone modification patterns determine the activity of the genomic regions associated with histones. In prostate cancer, deregulated AR interaction with its coregulators in the nucleus is frequently observed [11,15]; these interactions with coregulators and histone-modifying enzymes provoke AR-dependent epigenetic regulation [8,11]. AR enhancers are characterized by the presence of H3K4me1, H3K27ac, and H2A.Zac, which activate AR-enhancer function of RNA transcription [49]. Notably, histone modification patterns of AR binding regions are important for transcription of the AR-regulated genes involved in cancer progression [1,5].
The acetylation of histone proteins opens up the nucleosome packing within chromatin and increases the accessibility of chromatin remodeling factors to DNA, resulting in enhanced transcriptional activity [8,10,48,50]. The bromodomain proteins (BRDs) have the ability to recognize acetylated lysine residues [50]. This activity allows BRDs to play a critical role in histone-acetylation-mediated regulatory mechanisms for transcription in the chromatin. In addition, BRDs have been found to interact with AR via the bromodomain and are responsible for AR signaling. Aberrant expression of BRDs has been shown in prostate cancer tissues [51] and deregulation of AR and BRDs mediate global chromatin accessibility modulation for regulating the transcriptional profile. Such BRDs include the ATPase Family AAA Domain Containing 2 (ATAD2), BRD2, and Tripartite motif-containing 24 (TRIM24), all of which interact with AR and are highly expressed in CRPC [51,52]. Other studies revealed that the gene encoding an E3 ubiquitin ligase, known as substrate binding adaptor speckle-type POZ protein (SPOP), is the gene most mutated in many prostate cancer tissues; mutated SPOP enhances the expression level of BRD4 protein through the loss of binding to, and inducing the ubiquitination of BRD4 for, degradation [53]. Thus, expression and regulatory mechanisms of these BRD-containing proteins possess the potential for use as therapeutic targets in prostate cancer.
5. Protein-Protein Interaction Associated With Epigenetic Modifications
AR enhances promoter activity by direct or indirect interaction with many coregulators, such as steroid receptor co-activators (SRCs) and other histone-modifying enzymes [9]. H3K4 methylations (mono-, di- or tri-methylation) result in activation of the promoter and enhancer regions [10]. Several studies indicate that SET1/MLL histone methyltransferase is a key enzyme that induces histone H3K4 methylation in AR signaling [54,55,56]. Menin protein was shown to associate with the N-terminus of MLL and promote MLL-target gene expression [54]. In CRPC tumors, menin is highly expressed and involved in tumor growth by directly binding to AR for the recruitment of the MLL complex. Interestingly, small molecule inhibitors to reduce the menin-MLL interaction could potentially provide efficacious CRPC treatment. MLL complex activity is positively regulated to promote androgen-mediated gene induction. After androgen treatment, protein kinase C-related kinase 1 (PRK1) increased the threonine 11 phosphorylation (H3T11P) levels of H3 that is located near AR binding regions [55]. WD repeat-containing protein 5 (WDR5), a subunit of the SET1/MLL complex, is recruited to and interacts with H3T11P, and then it enhances the association of the MLL complex with AR for H3K4 tri-methylation (H3K4me3) activity [56]. Thus, WDR5 is an important epigenetic integrator.
In another study, PRK1-mediated phosphorylation was shown to be responsible for androgen-mediated reduction of H3K9 methylation, a histone marker of transcriptional silencing, by jumonji C domain-containing protein (JMJD2C) in cooperation with lysine-specific demethylase 1 (LSD1) [55,57]. However, LSD1 can also demethylate both mono- and di-methylated H3K4 in other studies [58]. In prostate cancer cells, protein kinase C beta 1 (PKCβ1) promotes histone H3T6 phosphorylation in an androgen-dependent manner by interacting with AR [59]. Notably, this phosphorylation event is a critical histone modification for specifically preventing demethylases from acting on H3K4 in ARBSs (Figure 1). Taken together, histone methylation through LSD1 and the MLL complex has an important role in AR-mediated transcriptional regulation.
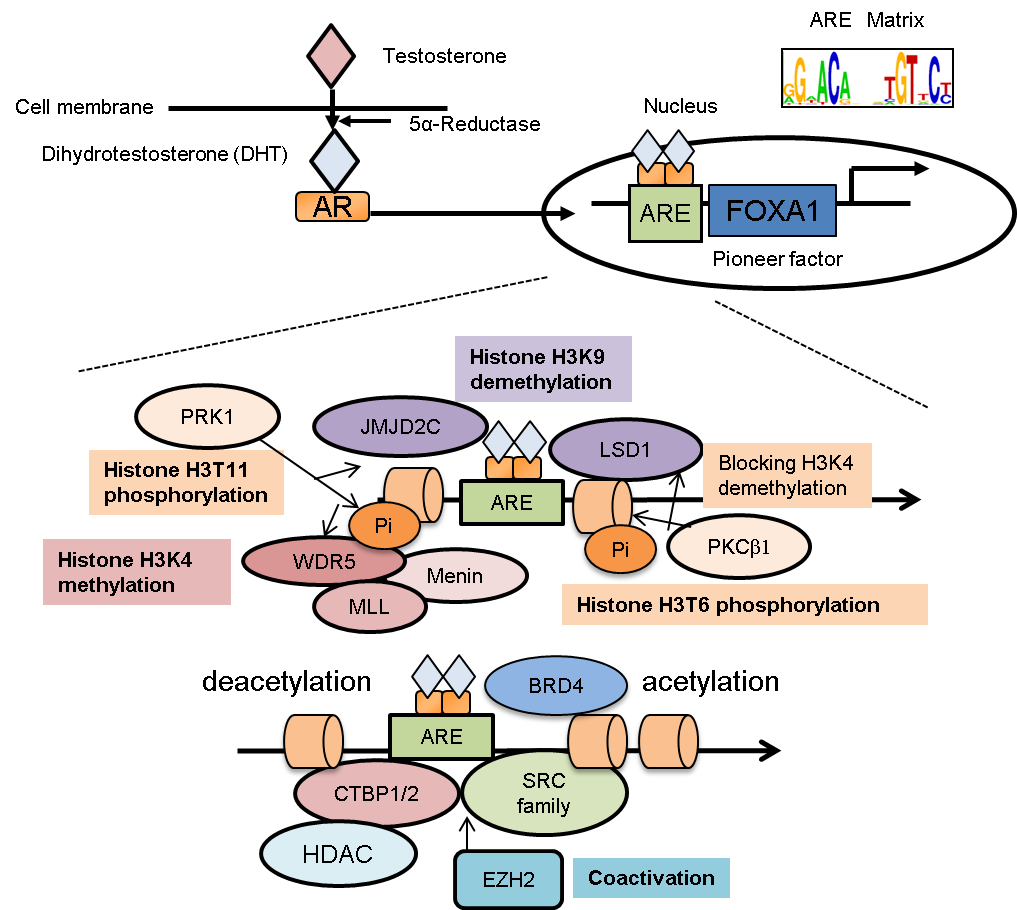
Figure 1 Epigenetic program in AR-binding regions. Androgen receptor (AR) translocates into the nucleus after androgen treatment. By collaborating with FOXA1, AR is recruited to specific loci known as androgen responsive elements (AREs). Then AR activates its target genes by modifying the epigenetic status around the AR-binding regions. As cofactors of AR, several histone-modifying enzymes are recruited to AR-binding sites. (1) PKCβ1-mediated histone (H3T6) phosphorylation directs LSD1 for H3K9 demethylation by cooperating with JMJD2C without affecting H3K4 methylation. (2) H3T11 phosphorylation accelerates both WDR5-mediated MLL recruitments and JMJD2C-mediated demethylation activity. MLL complex interacts with AR through menin and promotes histone H3K4 methylation to enhance AR-dependent gene expression. (3) For histone acetylation or deacetylation, the SRC family and CTBP1/2 are AR-interacting cofactors. (4) In CRPC, EZH2 functions as a coactivator that enhances AR-transcriptional activity, independent of its histone-methylation activity. (5) BRD proteins interact with histone-acetylated lysine and AR for recruitment of RNA pol II.
C-terminal binding protein 2 (CTBP2) is an androgen-responsive cofactor of AR [60]. CTBP2 repressed the transcription of tumor-suppressor genes and AR corepressors in prostate cancer cells. For example, nuclear receptor corepressor (NCOR) and receptor-interacting protein 140 (RIP140) are representative AR corepressors. CTBP2 is recruited to the AR-associated promoter or enhancer regions of these genes. Knockdown of CTBP2 increases the interaction of AR with these corepressors, which then decreases histone acetylation levels of ARBSs [60]. In addition, comprehensive gene-expression analyses indicate a positive role of CTBP2 in androgen-mediated gene regulation. Thus, targeting CTBP2 might be an effective treatment for CRPC by modulating epigenetic pathways associated with AR signaling.
6. Epigenetic and Post-Translational Regulatory Mechanisms for AR Overexpression and Activation
Several studies have shown that histone modification near the AR gene locus increased AR expression in CRPC [61,62]. Recruitment of AR and its associated cofactors such as LSD1 to the enhancer region situated in an intron of the AR gene represses the transcription of AR itself, by decreasing histone H3K4 methylation levels. This negative feedback mechanism controls AR expression in an androgen-dependent manner. Therefore, after long-term reduction of androgens to castration levels, AR expression is enhanced in prostate cancer cells. Interestingly, low concentrations of androgens can promote AR-mediated gene induction without repressing AR itself in CRPC cells [61]. Another study reported the importance of the AR enhancer, which is located 650 kb upstream from the AR transcriptional start site for AR overexpression in CRPC tissues [62]. Sequencing analysis revealed that multiple transacting factors, including HOXB13 and FOXA1, bind to this enhancer in prostate cancer tissues. Moreover, this enhancer region is amplified and epigenetically activated (histone H3K27 acetylation) in CRPC tissues, suggesting that this noncoding region functions as an oncogenic driver and becomes activated during the selective pressure exerted by androgen deprivation therapy [62]. Thus, epigenetic regulation of far-upstream and intragenic enhancers of the AR gene is an important mechanism for promoting the overexpression of AR.
Moreover, posttranslational modifications of AR lead to the enhancement of AR activity through regulation of protein stability, cellular localization, and structure during prostate cancer progression. Acetylation of AR is induced by androgen and cytokine treatments through acetylenes, such as p300 and p300/CREB binding protein (CBP)-associated factor (PCAF), and Tip60 [63,64,65]. Lysine residues (K630, K632, and K633) within the KLKK motif of the hinge region are targeted for acetylation. According to studies using mutants of AR, these acetylations have a role in the enhancement of the interaction with co-activators and genomic bindings. In addition, acetylated AR showed nuclear localization even in the absence of androgen, suggesting this modification is important for the cellular localization of AR [66]. In addition, dysregulation of AR phosphorylation has been implicated in CRPC development and well characterized in previous publications [67,68,69,70,71]. Ser81 was described as a representative phosphorylation site, and phosphorylation activity was increased by androgen treatment [67,68,69,70]. Protein kinase C (PKC), CDK1, CDK5, and CDK9 have been reported to be responsible for phosphorylated Ser81. This modification, occurring in the nucleus, enhances AR protein stability, chromatin binding, and promotes transcriptional activation [67,68,69]. In androgen-independent prostate cancer cells, increased levels of phosphorylated ser-81, along with AR stabilization has been observed [70]. Phosphorylation of Tyr534 has also been identified in AR. This phosphorylation is required for translocation to the nucleus and transcriptional response at low levels of androgen treatment. Tyr534 phosphorylation is also increased in CRPC samples [71]. Other modifications of AR include regulation of methylation [72], ubiquitination [73], deubiquitinating [74] and sumoylation [75,76]; these modifications are important for increasing AR-protein stability and transcriptional activity [77]. These modifications and the enzymes responsible for the modification of the receptor are critical candidates for a novel therapeutic strategy.
7. Development of Drugs Targeting Epigenetic Pathways in Prostate Cancer
Recently, BET bromodomain protein inhibitors have been shown to be a novel approach for epigenetically treating CRPC [78,79,80]. In these reports, a representative BET bromodomain inhibitor, JQ1, promoted apoptosis, and inhibited AR-target-gene expression in prostate cancer cells [79,80]. JQ1 weakened the BRD4-AR bond, resulting in RNA polymerase II detaching from the AR binding regions [79]. BET inhibitors have strong in vivo efficacy in both CRPC xenograft and patient-derived models [79]. Clinical trials in prostate cancer patients are ongoing for several BET inhibitors. Thus, these results indicate that modulation of AR epigenetic function through BET proteins could be useful in treating prostate cancer.
Global analysis of polycomb repressive complex 2 (PRC2) determined that it represses by epigenetic function the expression of tumor-suppressive genes in prostate cancer [31,81]. A PRC2 component, enhancer of zeste 2 (EZH2), trimethylates histone H3 lysine 27 (H3K27) to suppress gene expression and is overexpressed in metastatic CRPC tissues [82]. In addition, another report suggested that EZH2 functions as a co-activator to activate transcription factors such as AR in CRPC [81]. Co-activator function requires phosphorylation of EZH2 and a methyltransferase domain is involved in the process [81]. Recently, selective EZH2 inhibitors such as GSK126 have been identified, which have anti-proliferative activity in prostate cancer cells [83]. Embryonic ectoderm development (EED) is another main component of the PRC2 complex; its inhibitor, MAK683, has recently entered a clinical trial (Phase I) that includes prostate cancer patients [84]. Thus, these studies raise the possibility that inhibitors of PRC2 could effectively treat CRPC.
Because LSD1 controls androgen-dependent prostate cancer cell proliferation by regulating AR activity through histone modifications as described above, several reports have focused on drugs targeting LSD1 [85,86]. By in vitro screening using protein-structure similarity research, Namoline, a γ-pyrone, was identified as a reversible, selective, and novel LSD1 inhibitor [85]. In this report, Namoline treatment reduced H3K9me2 levels, a histone marker for LSD1 activity, in LNCaP cells. A blockade of LSD1 by Namoline silenced expression of genes regulated by AR and then severely abrogated androgen-dependent cell growth [85]. In addition, another study reveals that LSD1 facilitates the survival of prostate cancer cells, including in CRPC, independently of both its demethylase function and of AR [86]. LSD1 promotes prostate cancer gene expression in collaboration with other transcription factors, such as ZNF217, independently of AR [86]. The small-molecule LSD1 inhibitor, SP-2509, blocks important demethylase-independent functions and suppresses CRPC cell viability, suggesting the potential benefit of LSD1 inhibition in this disease [86]. Thus, demethylation-dependent and -independent pathways of LSD1 could have important roles in prostate cancer progression.
Histone deacetylase (HDAC) inhibitors have been shown to suppress AR activity in a recent report [87]. Mechanistically, decreased AR activity is caused by miRNA-mediated suppression (post-transcriptional regulation). Among miRNAs induced by HDAC inhibitor treatment, miR-320 was identified to be closely involved in modifying AR expression. Interestingly, low expression of miR-320 was significantly associated with poor prognosis of prostate cancer patients. Treatment with an miR-320a mimic decreased AR protein expression and suppressed tumor growth [88]. Thus, the miRNA-mediated epigenetic pathway may have potential as a developing therapy against CRPC.
Another major AR co-activator, CREB-binding protein (CBP)/p300, is also an important histone acetyltransferase to enhance AR transcriptional activity [89,90,91]. CBP/p300 is shown to have an oncogenic function and is upregulated in tumor compared to benign tissues [89]. However, currently available inhibitors targeting the histone acetyltransferase domain within CBP/p300 (e.g., natural products, bi-substrate analogues, and the small molecule C6466) lack selectivity and potency. Recently, two papers reported the development of a new CBP/p300 selective inhibitor [90,91], which inhibited proliferation of several cancer cells, including AR-positive prostate cancer [90,91]. Both inhibitors decreased AR signaling in both androgen-dependent prostate cancer and CRPC and inhibited tumor growth in a patient-derived, castration-resistant xenograft model. Thus, these reports showed that selective and potent small molecule inhibitors targeting the catalytic activity of histone acetyltransferases could be effective drugs for treating CRPC.
8. The Role of Non-Coding RNAs in the Biology of Prostate Cancer and AR Signals
DNA sequence techniques, which have rapidly developed, have shown that more than 90% of the human genome is transcribed actively, but only a small portion (~ 2%) of these transcripts is translated into proteins [92]. Most of the transcripts expressed in the nucleus are non-coding RNAs (ncRNAs), which were initially considered “dark matter.” NcRNAs are mainly categorized into two types, short and long. Short, non-coding RNAs are up to 200 nucleotides in length and include transfer RNA, microRNA (miRNA), snoRNA, and enhancer-templated non-coding RNAs (eRNAs) [93]. By post-transcriptionally modulating their target mRNA or protein expression, miRNAs actively function in cancer progression. LncRNAs compose the majority of the expressed ncRNAs in the human genome and are longer than 200 nucleotides [94].
In general, miRNAs associate with the 3’ untranslated region (UTR) of the target mRNAs to repress their translation. Several studies have analyzed miRNA expression patterns in prostate cancer tissues and observed that specific miRNAs were up- or down-regulated during the disease progression [95,96,97]. Global analysis of miRNAs in prostate cancer cells revealed that miR-21, miR-29a/b, miR-32, miR-125b, miR-141, miR-148a, and miR-200a were AR-regulated miRNAs, and all have clinical relevance in prostate cancer [95,96,97,98,99]. Many experimental results have demonstrated that these AR-regulated miRNAs promote prostate tumor growth by regulating cell cycle, apoptosis, and microenvironment [92].
Furthermore, some miRNAs regulate AR-mediated epigenetic function by targeting AR or AR-associated factors. For instance, androgen-inducible miR-141 correlates with the development of prostate cancer [94,98] and the orphan receptor small heterodimer partner (SHP) represses AR-mediated transcription, functioning as a corepressor [100]. Interestingly, miR-141 binds to the 3’ UTR of SHP mRNA and inhibits the SHP mRNA expression, leading to the enhancement of AR transcriptional activity. In another study, repression of 12 miRNAs expression levels (miR-1, miR-133a, miR-133b, miR-135a, miR-143-3p, miR-145-3p, miR-205, miR-221-3p, miR-221-5p, miR-222-3p, miR-24-1-5p, and miR-31) was demonstrated in metastatic prostate cancer tissues [101]. Notably, these miRNAs target AR and members of the steroid-receptor co-activator family (SRC family). This study showed that miR-135a is transcriptionally induced by androgen treatment and that AR was recruited to the miR-135a locus [101]. Therefore, androgen-deprivation therapy enhances AR expression by silencing this miRNA in prostate cancer. Moreover, miR-205 is dysregulated in prostate cancer compared to benign tissues. Clinical studies revealed that the miR-205 expression level in cancer is inversely correlated with AR and the prognosis of patients, suggesting that AR is a direct target of this miRNA [102]. In addition, the miR-205 expression level in CRPC tissues was lower than what was found in patients who had not been treated with anti-androgen therapy [103]. Thus, it is possible that reduction of these miRNAs’ expression will be a key event for CRPC development and progression.
Interestingly, a recent study has shown that several miRNAs, such as the miR-29 family and miR-22, are highly regulated by androgen in anti-AR therapy-refractory prostate cancer [104]. In prostate cancer tissues, the expression level of miR-29a/b was found to be inversely correlated with that of ten-eleven translocation 2 (TET2). The TET family of proteins catalyze the production of 5-hydroxymethylcytosine (5-hmC) from 5-mC. Notably, 5-hmC functions as a stable epigenetic marker as well as an intermediate product of a demethylation process [105]. Mechanistically, TET2 repression by miRNAs inhibits 5-hmC modifications; this inhibition then promotes FOXA1-binding enhancer activity (Figure 2). FOXA1 is responsible for the induction of prostate cancer-related gene expression. Interestingly, a representative 5-hmC-regulated gene was mTOR, a mammalian target of rapamycin. These experimental and clinical results are consistent with other reports of miR-29 family [106,107]. Exome sequencing of the genomes derived from CRPC samples demonstrated somatic mutations within the TET2 exon [108]; TET2 could also directly regulate AR signaling by binding to AR [109]. Taken together, these reports suggest that androgen-regulated miRNAs modulate the 5-hmC epigenetic conditions by TET2 regulation in prostate cancer, particularly in a subset of metastatic disease.
Global analysis of RNA production in prostate cancer cells revealed that androgen treatment induced eRNA expression in ARBSs (Figure 2). eRNA contributes to nucleosome remodeling and induces the interaction between enhancer and promoter by forming a loop to promote gene activation [110]. Recruitment of AR was widely observed in these enhancer/promoter regions occupied with eRNAs [111]. Interestingly, silencing eRNAs inhibits androgen-dependent gene induction through enhancer-activity regulation [111]. DNA-nicking activity of topoisomerase I (TOP1) was shown to induce eRNA expression, which activates enhancers [112]; furthermore, kinetic recruitment of DNA-damage-repair machinery to the ARBSs modulates nucleosome remodeling [112]. Thus, these studies highlight the importance of eRNA production in AR-mediated nucleosome remodeling and epigenetic regulation.
9. LncRNA: Epigenetic Regulators in AR-Binding Enhancer Regions
The structure and biogenesis of lncRNAs are similar to those of mRNAs in that lncRNAs are polyadenylated and can function in either the nuclear or the cytoplasmic compartments [113]. In addition, lncRNAs can be folded into secondary and tertiary structures to exert their functions. Growing evidence has shown that lncRNAs are involved in many human diseases, such as cancer [113,114], and reports recently showed that AR epigenetic function could be directly regulated by lncRNA [115,116,117,118]. Steroid receptor RNA activator (SRA) was shown to modulate, through interaction, the transcriptional activity of nuclear receptors including AR, estrogen receptor (ER), progesterone receptor (PR), glucocorticoid receptor (GR), and thyroid hormone receptor (TR) [115], providing the first evidence that lncRNA is associated with nuclear receptor function. Mechanistically, SRA interacts with steroid receptor co-activator (SRC-1), a representative co-activator for nuclear receptors and six stem-loop motifs found in the sequence of SRA are responsible for the activation of co-activators. Notably, SRA-expression levels were shown to be elevated in many tumor tissues, including prostate cancer, compared to normal tissues [115,116].
Two prostate-cancer-related lncRNAs, prostate cancer gene expression marker 1 (PCGEM1) and prostate cancer noncoding RNA 1 (PRNCR1), were shown to cooperatively function in AR gene regulation [118]. PCGEM1 was identified as an androgen-regulated lncRNA that is specifically found in prostate cancer tissues [119], and is highly expressed in prostate tumors and involved in the cellular anti-apoptotic activity by repressing the expression of p53 and p21 [120]. PRNCR1 locates to the vicinity of single nucleotide polymorphisms (SNPs) associated with prostate cancer susceptibility [121]. Importantly, both PCGEM1 and PRNCR1 interact with AR to promote AR activation [118]. In addition, PCGEM1 interacts with pygopus homolog 2 and PRNCR1 with DOT1-like histone H3 methyltransferase to modulate the AR protein post-translationally. These two enzymatic events then induce chromatin remodeling and the AR-occupied loop formation between enhancer and promoter (Figure 2). Suppressor of cytokine signaling 2-antisense transcript 1, another lncRNA associated with AR, was reported to be involved in AR activation. It promotes the recruitment of coregulators, like CTBP2, to ARBSs for epigenetic control [122]. Thus, these lncRNAs affect AR-mediated transcriptional activation by modulating epigenetic and chromatin structure.
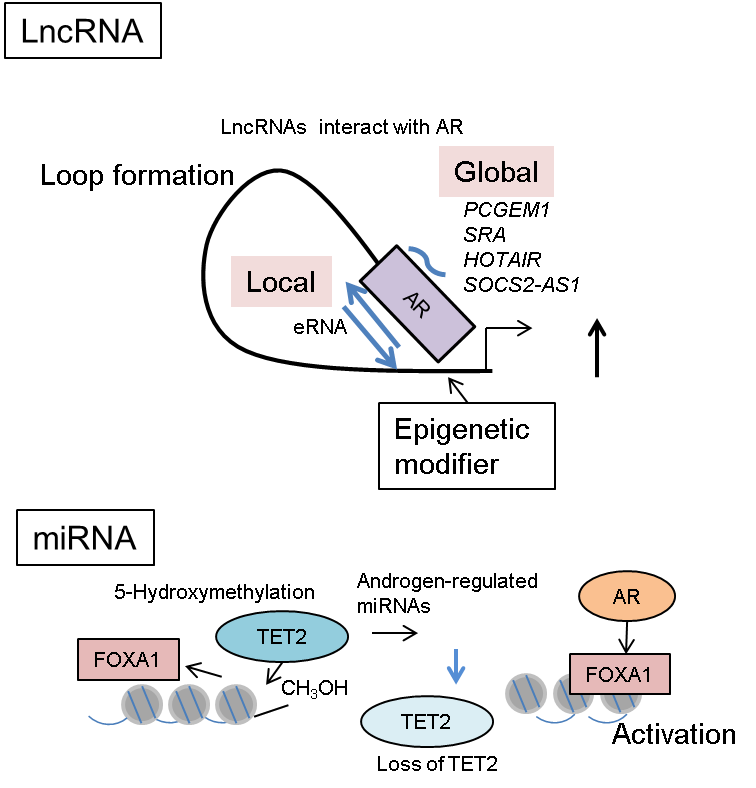
Figure 2 The role of non-coding RNA in AR-mediated epigenetic regulation. The functions of enhancer RNA (eRNA) expressed in AR-occupied enhancers or other AR-interacting lncRNAs from distant regions. These lncRNAs promote loop formation for promoter/enhancer interaction, including AR-binding sites and activate transcription. As for miRNAs, TET2 repression by androgen-regulated miRNAs inhibits 5-hmC modifications in FOXA1-associated enhancer regions. Removal of 5-hmC modification from DNA enhances the transcription regulated by FOXA1 and AR. Thus, 5-hmC functions as an epigenetic mark for repressing global FOXA1-mediated enhancers.
HOX antisense Intergenic RNA (HOTAIR) is transcribed in the antisense region of the HOXC gene cluster [123,124]. By associating with the polycomb repressive complex 2 (PRC2), HOTAIR acts as a transcriptional regulator. Moreover, HOTAIR also interacts with the LSD1/CoREST/REST complex. This association couples PRC2 and LSD1 to enhance histone H3K27 methylation and K4 demethylation levels for gene repression [117]. Thus, interaction of HOTAIR with chromatin-remodeling factors increased the formation heterochromatin at specific regions important for gene regulation. Moreover, high expression of HOTAIR is significantly associated with a poor prognosis of breast and prostate cancer patients [123,125]. This result is consistent with the positive regulation of AR by HOTAIR [125]. HOTAIR is repressed by androgen treatment and overexpressed by androgen deprivation. Interestingly, HOTAIR binds with AR and together they inhibit the function of MDM2, an E3-ubiquitin ligase, preventing the degradation of the AR protein. This interaction between HOTAIR and AR blocks ubiquitin-mediated degradation by MDM2 and therefore stabilizes AR protein levels to enhance AR downstream signals for promoting CRPC [125]. Taken together, the results of this study highlight the role of lncRNA for post-translational regulation of AR to enhance AR signaling.
Recent sequencing studies have demonstrated active transcription from both sense and antisense strands in the human genome [92,114,126]. More than 1,000 pairs of sense-antisense transcripts were found, suggesting that bidirectional gene regulation takes place widely throughout the entire genome [114]. Comprehensive analysis of androgen-regulated transcripts by cap analysis of gene expression found a novel androgen-responsive antisense RNA, CTBP1-AS [126]. C-terminal binding protein 1 (CTBP1) is a negative regulator of AR signals by ligand-dependent interaction with AR. Direct binding of AR to the promoter of CTBP1-AS induced its transcription. To regulate the expression of its target gene, CTBP1-AS physically interacts with PTB-associated splicing factor (PSF), which is a DNA/RNA binding protein [126,127,128]. Because PSF functions as a transcriptional regulator by binding to DNA and associating with histone deacetylase (HDAC) complexes, CTBP1-AS-bound PSF transcriptionally represses its target genes, including CTBP1 [126]. Moreover, CTBP1-AS induces cell cycle progression by transcriptionally repressing specific cell cycle inhibitors such as p53 and SMAD3 in prostate cancer cells [99]. Thus, androgen-induced CTBP1-AS mediates the AR epigenetic regulation of gene expression and promotes prostate-cancer associated signals. These results highlight the new role of antisense RNAs and RNA-binding proteins in cancer progression and target these pathways as potentially useful therapeutic strategies against CRPC.
Interestingly, another report has shown that PSF enhances the stability of another CRPC-related lncRNA, second chromosome locus associated with prostate 1 (SChLAP1) [129], and the mRNAs of AR-target signals, by direct interactions [127]. SChLAP1 is overexpressed in prostate cancers and is a predictor of metastasis and mortality [129]. It was demonstrated that SChLAP1 interacts with chromatin complex SWI/SNF and inhibits the function of SNF5, an important component of this complex [129]; this suggests that SChLAP1 inhibits tumor-suppressive functions of the SWI/SNF complex; this leads to gene-expression modulation. More importantly, pathway analysis showed that PSF primarily targets spliceosome genes in CRPC cells to enhance their expressions [127]. Interestingly, in addition to PSF, the wide range of spliceosome genes are overexpressed in metastatic prostate cancer tissues, suggesting the importance of splicing factors in the disease progression. PSF also binds to AR mRNA, facilitating splicing of AR to enable the production of AR-V7 in CRPC cells [127]. Heterogeneous nuclear ribonucleoprotein L (HNRNPL) was also identified to be required for prostate cancer growth by a CRISPR/Cas9 knockout screen. HNRNPL regulates the alternative splicing of a set of RNAs, which includes the AR transcript [130]. Thus, the RNA-binding proteins play an important role in modulating lncRNA function, as well as in regulating prostate-cancer-associated gene expression such as AR, for driving prostate cancer progression.
10. Conclusion
AR signaling drives the majority of prostate cancer and CRPC tumor development. Resistance to hormone therapy can be acquired through multiple mechanisms for AR activation. AR coregulators and collaborating the transcription factors are essential for AR to exert its epigenetic and transcriptional activity. The role of non-coding RNAs, such as lncRNAs and miRNAs, has been shown to be critical in AR-mediated epigenetic machinery. Although AR-targeting drugs have been developed, they cannot eliminate CRPC due to the adaptive evolution of the disease during the treatment; therefore, drugs targeting epigenetic factors involved in AR signaling are promising therapies for treating prostate cancer and CRPC.
Acknowledgments
This work was supported by grants from the JSPS (number 17H04334), Japan; grants from Takeda Science Foundation, Japan.
Author Contributions
The author made all contributions to this work.
Competing Interests
The authors have declared that no competing interests exist.
References
- Takayama K, Inoue S. Transcriptional network of androgen receptor in prostate cancer progression. Int J Urol. 2013; 20: 756-768. [CrossRef]
- Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2014; 33: 2815-2825. [CrossRef]
- Attard G, Parker C, Eeles RA, Schröder F, Tomlins SA, Tannock I, et al. Prostate cancer. Lancet 2016; 387: 70-82. [CrossRef]
- Bill-Axelson A, Holmberg L, Ruutu M, Garmo H, Stark JR, Busch C, et al. Radical prostatectomy or watchful waiting in early prostate cancer. N. Engl. J. Med. 2014; 370: 932-942. [CrossRef]
- Takayama K. The biological and clinical advances of androgen receptor function in age-related diseases and cancer. Endocr J. 2017; 64: 933-946. [CrossRef]
- Chang C, Yeh S, Lee SO, Chang TM. Androgen receptor (AR) pathophysiological roles in androgen-related diseases in skin, bone/muscle, metabolic syndrome and neuron/immune systems: lessons learned from mice lacking AR in specific cells. Nucl Recept Signal. 2013; 11: e001. [CrossRef]
- Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activatorsmediates binding to nuclear receptors. Nature. 1997; 387: 733-736. [CrossRef]
- Cucchiara V, Yang JC, Mirone V, Gao AC, Rosenfeld MG, Evans CP. Epigenomic regulation of androgen receptor signaling: potential role in prostate cancer therapy. Cancers (Basel). 2017; 9. [CrossRef]
- Shang Y Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell. 2002; 9: 601-610. [CrossRef]
- Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008; 132: 958-970. [CrossRef]
- Perissi V, Rosenfeld MG. Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol. 2005; 6: 542-554. [CrossRef]
- Jenster G, van der Korput HA, van Vroonhoven C, van der Kwast TH, Trapman J, Brinkmann AO. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol. 1991; 5: 1396-1404. [CrossRef]
- Jenster G, van der Korput HA, Trapman J, Brinkmann AO. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem. 1995; 270: 7341-7346. [CrossRef]
- Dehm SM, Regan KM, Schmidt LJ, Tindall DJ. Selective role of an NH2-terminal WxxLF motif for aberrant androgen receptor activation in androgen depletion-independent prostate cancer cells. Cancer Res. 2007; 67: 10067-10077. [CrossRef]
- Groner AC, Brown M. Role of steroid receptor and coregulator mutations in hormone-dependent cancers. J Clin Invest. 2017; 127: 1126-1135. [CrossRef]
- Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995; 332: 1393-1398. [CrossRef]
- Cai C, Balk SP. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr Relat Cancer. 2011; 18: 175-182. [CrossRef]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004; 10: 33-39. [CrossRef]
- de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011; 364: 1995-2005. [CrossRef]
- Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009; 324: 787-790. [CrossRef]
- Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012; 367: 1187-1197. [CrossRef]
- Culig Z. Molecular mechanisms of enzalutamide resistance in prostate cancer. Curr Mol Biol Rep. 2017; 3: 230-235. [CrossRef]
- Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. 2018; 378: 1408-1418. [CrossRef]
- Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010; 120: 2715-2730. [CrossRef]
- Waltering KK, Helenius MA, Sahu B, Manni V, Linja MJ, Jänne OA, et al. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009; 69: 8141-8149. [CrossRef]
- He Y, Lu J, Ye Z, Hao S, Wang L, Kohli M, et al. Androgen receptor splice variants bind to constitutively open chromatin and promote abiraterone-resistant growth of prostate cancer. Nucleic Acids Res. 2018; 46: 1895-1911. [CrossRef]
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014; 371: 1028-1038. [CrossRef]
- Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem. 2012; 287: 19736-19749. [CrossRef]
- Yang YC, Banuelos CA, Mawji NR, Wang J, Kato M, Haile S, et al. Targeting androgen receptor activation function-1 with EPI to overcome resistance mechanisms in castration-resistant prostate cancer. Clin Cancer Res. 2016; 22: 4466-4477. [CrossRef]
- Results from the phase 1 clinical trial of EPI-506 for treatment of mCRPC and updates clinical and strategic plans. ESSA Pharma. 2017. Available from: http://www.essapharma.com/wp-content/uploads/2017/09/BRANDED_ESSA-Press-Release-11Sept2017_FINAL.pdf.
- Cao Q, Mani RS, Ateeq B, Dhanasekaran SM, Asangani IA, Prensner JR et al. Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell. 2011; 20: 187-199. [CrossRef]
- Takayama K, Tsutsumi S, Suzuki T, Horie-Inoue K, Ikeda K, Kaneshiro K, et al. Amyloid precursor protein is a primary androgen target gene that promotes prostate cancer growth. Cancer Res. 2009; 69: 137-142. [CrossRef]
- Ashikari D, Takayama K, Obinata D, Takahashi S, Inoue S. CLDN8, an androgen-regulated gene, promotes prostate cancer cell proliferation and migration. Cancer Sci. 2017; 108: 1386-1393. [CrossRef]
- Takayama K, Horie-Inoue K, Suzuki T, Urano T, Ikeda K, Fujimura T, et al. TACC2 is an androgen-responsive cell cycle regulator promoting androgen-mediated and castration-resistant growth of prostate cancer. Mol Endocrinol. 2012; 26: 748-761. [CrossRef]
- Ashikari D, Takayama K, Tanaka T, Suzuki Y, Obinata D, Fujimura T, et al. Androgen induces G3BP2 and SUMO-mediated p53 nuclear export in prostate cancer. Oncogene. 2017; 36: 6272-6281. [CrossRef]
- Takayama K, Suzuki T, Tanaka T, Fujimura T, Takahashi S, Urano T, et al. TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene. 2018; 37: 2165-2180. [CrossRef]
- Takayama K, Suzuki T, Fujimura T, Takahashi S, Inoue S. Association of USP10 with G3BP2 inhibits p53 signaling and contributes to poor outcome in prostate cancer. Mol Cancer Res. 2018; 16: 846-856. [CrossRef]
- Takayama K, Suzuki T, Fujimura T, Takahashi S, Inoue S. COBLL1 modulates cell morphology and facilitates androgen receptor genomic binding in advanced prostate cancer. Proc Natl Acad Sci U S A. 2018; 115: 4975-4980. [CrossRef]
- Takayama K, Suzuki T, Tsutsumi S, Fujimura T, Takahashi S, Homma Y, et al. Integrative analysis of FOXP1 function reveals a tumor-suppressive effect in prostate cancer. Mol Endocrinol. 2014; 28: 2012-2024. [CrossRef]
- Urbanucci A, Sahu B, Seppälä J, Larjo A, Latonen LM, Waltering KK, et al. Overexpression of androgen receptor enhances the binding of the receptor to the chromatin in prostate cancer. Oncogene. 2012; 31: 2153-2163. [CrossRef]
- Wang Q, Li W, Liu XS, Carroll JS, Jänne OA, Keeton EK, et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007; 27: 380-392. [CrossRef]
- Obinata D, Takayama K, Fujiwara K, Suzuki T, Tsutsumi S, Fukuda N, et al. Targeting Oct1 genomic function inhibits androgen receptor signaling and castration-resistant prostate cancer growth. Oncogene. 2016; 35: 6350-6358. [CrossRef]
- Takayama K, Suzuki T, Tsutsumi S, Fujimura T, Urano T, Takahashi S, et al. RUNX1, an androgen- and EZH2-regulated gene, has differential roles in AR-dependent and -independent prostate cancer. Oncotarget. 2015; 6: 2263-2276. [CrossRef]
- Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013; 23: 35-47. [CrossRef]
- Pomerantz MM, Li F, Takeda DY, Lenci R, Chonkar A, Chabot M, et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Genet. 2015; 47: 1346-1351. [CrossRef]
- Long MD, Smiraglia DJ, Campbell MJ. The genomic impact of DNA CpG methylation on gene expression; relationships in prostate cancer. Biomolecules. 2017; 7: E15. [CrossRef]
- Castillo-Aguilera O, Depreux P, Halby L, Arimondo PB, Goossens L. DNA methylation targeting: The DNMT/HMT crosstalk challenge. Biomolecules. 2017; 7: E3. [CrossRef]
- McAnena P, Brown JA, Kerin MJ. Circulating nucleosomes and nucleosome modifications as biomarkers in cancer. Cancers (Basel). 2017; 9: E5. [CrossRef]
- Valdés-Mora F, Gould CM, Colino-Sanguino Y, Qu W, Song JZ, Taylor KM, et al. Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat Commun. 2017; 8: 1346. [CrossRef]
- Sanchez R, Meslamani J, Zhou MM. The bromodomain: from epigenome reader to druggable target. Biochim Biophys Acta. 2014; 1839: 676-685. [CrossRef]
- Urbanucci A, Barfeld SJ, Kytölä V, Itkonen HM, Coleman IM, Vodák D et al. Androgen receptor deregulation drives bromodomain-mediated chromatin alterations in prostate cancer. Cell Rep. 2017; 19: 2045-2059. [CrossRef]
- Groner AC, Cato L, de Tribolet-Hardy J, Bernasocchi T, Janouskova H, Melchers D, et al. TRIM24 Is an Oncogenic transcriptional activator in prostate cancer. Cancer Cell. 2016; 29: 846-858. [CrossRef]
- Zhang P, Wang D, Zhao Y, Ren S, Gao K, Ye Z, et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat Med. 2017; 23: 1055-1062. [CrossRef]
- Malik R, Khan AP, Asangani IA, Cieślik M, Prensner JR, Wang X, et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat Med. 2015; 21: 344-352. [CrossRef]
- Metzger E, Yin N, Wissmann M, Kunowska N, Fischer K, Friedrichs N, et al. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat Cell Biol. 2008; 10: 53-60. [CrossRef]
- Kim JY, Banerjee T, Vinckevicius A, Luo Q, Parker JB, Baker MR, et al. A role for WDR5 in integrating threonine 11 phosphorylation to lysine 4 methylation on histone H3 during androgen signaling and in prostate cancer. Mol Cell. 2014; 54: 613-625. [CrossRef]
- Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005; 437: 436-439. [CrossRef]
- Yang M, Gocke CB, Luo X, Borek D, Tomchick DR, Machius M, et al. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol Cell. 2006; 23: 377-387. [CrossRef]
- Metzger E, Imhof A, Patel D, Kahl P, Hoffmeyer K, Friedrichs N, et al. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature. 2010; 464: 792-796. [CrossRef]
- Takayama K, Suzuki T, Fujimura T, Urano T, Takahashi S, Homma Y, et al. CtBP2 modulates the androgen receptor to promote prostate cancer progression. Cancer Res. 2014; 74: 6542-6553. [CrossRef]
- Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011; 20: 457-471. [CrossRef]
- Takeda DY, Spisák S, Seo JH, Bell C, O'Connor E, Korthauer K, et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell. 2018; 174: 422-432. [CrossRef]
- Fu M, Wang C, Reutens AT, Wang J, Angeletti RH, Siconolfi-Baez L, et al. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J Biol Chem. 2000; 275: 20853-20860. [CrossRef]
- Lee SO, Chun JY, Nadiminty N, Lou W, Feng S, Gao AC. Interleukin-4 activates androgen receptor through CBP/p300. Prostate. 2009; 69: 126-132. [CrossRef]
- Gaughan L, Logan IR, Cook S, Neal DE, Robson CN. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J Biol Chem. 2002; 277: 25904-25913. [CrossRef]
- Shiota M, Yokomizo A, Masubuchi D, Tada Y, Inokuchi J, Eto M, et al. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate. 2010; 70: 540-554.
- Chen S, Xu Y, Yuan X, Bubley GJ, Balk SP. Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin-dependent kinase 1. Proc Natl Acad Sci U S A. 2006; 103: 15969-15974. [CrossRef]
- Chen S, Gulla S, Cai C, Balk SP. Androgen receptor serine 81 phosphorylation mediates chromatin binding and transcriptional activation. J Biol Chem. 2012; 287: 8571-8583. [CrossRef]
- Hsu FN, Chen MC, Chiang MC, Lin E, Lee YT, Huang PH, et al. Regulation of androgen receptor and prostate cancer growth by cyclin-dependent kinase 5. J Biol Chem. 2011; 286: 33141-33149. [CrossRef]
- Gordon V, Bhadel S, Wunderlich W, Zhang J, Ficarro SB, et al. CDK9 regulates AR promoter selectivity and cell growth through serine 81 phosphorylation. Mol Endocrinol. 2010; 24: 2267-2280. [CrossRef]
- Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005; 8: 393-406. [CrossRef]
- Ko S, Ahn J, Song CS, Kim S, Knapczyk-Stwora K, Chatterjee B. Lysine methylation and functional modulation of androgen receptor by Set9 methyltransferase. Mol Endocrinol. 2011; 25: 433-444. [CrossRef]
- Gaughan L, Logan IR, Neal DE, Robson CN. Regulation of androgen receptor and histone deacetylase 1 by Mdm2-mediated ubiquitylation. Nucleic Acids Res. 2005; 33: 13-26. [CrossRef]
- Faus H, Meyer HA, Huber M, Bahr I, Haendler B. The ubiquitin-specific protease USP10 modulates androgen receptor function. Mol Cell Endocrinol. 2005; 245: 138-146. [CrossRef]
- Toropainen S, Malinen M, Kaikkonen S, Rytinki M, Jääskeläinen T, Sahu B, et al. SUMO ligase PIAS1 functions as a target gene selective androgen receptor coregulator on prostate cancer cell chromatin. Nucleic Acids Res. 2015; 43: 848-861. [CrossRef]
- Nishida T, Yasuda H. PIAS1 and PIASxalpha function as SUMO-E3 ligases toward androgen receptor and repress androgen receptor-dependent transcription. J Biol Chem. 2002; 277: 41311-41317. [CrossRef]
- Coffey K, Robson CN. Regulation of the androgen receptor by post-translational modifications. J Endocrinol. 2012; 215: 221-237. [CrossRef]
- Sanchez R, Meslamani J, Zhou MM. The bromodomain: from epigenome reader to druggable target. Biochim Biophys Acta. 2014; 1839: 676-685. [CrossRef]
- Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014; 510: 278-282. [CrossRef]
- Wyce A, Degenhardt Y, Bai Y, Le B, Korenchuk S, Crouthame MC, et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget. 2013; 4: 2419. [CrossRef]
- Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012; 338: 1465-1469. [CrossRef]
- Ren G, Baritaki S, Marathe H, Feng J, Park S, Beach S, et al. Polycomb protein EZH2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor RKIP in breast and prostate cancer. Cancer Res. 2012; 72: 3091-3104. [CrossRef]
- Wu C, Jin X, Yang J, Yang Y, He Y, Ding L, et al. Inhibition of EZH2 by chemo- and radiotherapy agents and small molecule inhibitors induces cell death in castration-resistant prostate cancer. Oncotarget. 2016; 7: 3440-3452. [CrossRef]
- Baumgart SJ, Haendler B. Exploiting epigenetic alterations in prostate cancer. Int J Mol Sci. 2017; 18: E1017. [CrossRef]
- Willmann D1, Lim S, Wetzel S, Metzger E, Jandausch A, Wilk W, et al. Impairment of prostate cancer cell growth by a selective and reversible lysine-specific demethylase 1 inhibitor. Int J Cancer. 2012; 131: 2704-2709. [CrossRef]
- Sehrawat A, Gao L, Wang Y, Bankhead A 3rd, McWeeney SK, King CJ, et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc Natl Acad Sci U S A. 2018; 115: E4179-E4188.
- Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N, et al. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009; 69: 958-966. [CrossRef]
- Sato S, Katsushima K, Shinjo K, Hatanaka A, Ohka F, Suzuki S, et al. Histone deacetylase inhibition in prostate cancer triggers miR-320-mediated suppression of the androgen receptor. Cancer Res. 2016; 76: 4192-4204. [CrossRef]
- Linja MJ, Porkka KP, Kang Z, Savinainen KJ, Jänne OA, Tammela TL, et al. Expression of androgen receptor coregulators in prostate cancer. Clin Cancer Res. 2004; 10: 1032-1040. [CrossRef]
- Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017; 550: 128-132. [CrossRef]
- Jin L, Garcia J, Chan E, de la Cruz C, Segal E, Merchant M, et al. Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of castration-resistant prostate cancer. Cancer Res. 2017; 77: 5564-5575. [CrossRef]
- Takayama K, Inoue S. The emerging role of noncoding RNA in prostate cancer progression and its implication on diagnosis and treatment. Brief Funct Genomics. 2016; 15: 257-265. [CrossRef]
- Iadevaia V, Gerber AP. Combinatorial control of mRNA fates by RNA-binding proteins and non-coding RNAs. Biomolecules. 2015; 5: 2207-2222. [CrossRef]
- Takayama K, Misawa A, Inoue S, Significance of microRNAs in androgen signaling and prostate cancer progression, Cancers (Basel), 2017; 9: E102. [CrossRef]
- Ribas J, Ni X, Haffner M, Wentzel EA, Salmasi AH, Chowdhury WH, et al. miR-21: an androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res 2009; 69: 7165-7169. [CrossRef]
- Coppola V, Musumeci M, Patrizii M, Cannistraci A, Addario A, Maugeri-Saccà M, et al. BTG2 loss and miR-21 upregulation contribute to prostate cell transformation by inducing luminal markers expression and epithelial-mesenchymal transition. Oncogene. 2013; 32: 1843-1853. [CrossRef]
- Shi XB, Xue L, Yang J, Ma AH, Zhao J, Xu M, et al. An androgen-regulated miRNA suppresses Bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc Natl Acad Sci U S A. 2007; 104: 19983-19988. [CrossRef]
- Murata T, Takayama K, Katayama S, Urano T, Horie-Inoue K, Ikeda K, et al. miR-148a is an androgen-responsive microRNA that promotes LNCaP prostate cell growth by repressing its target CAND1 expression. Prostate Cancer Prostatic Dis. 2010; 13: 356-361. [CrossRef]
- Jalava SE, Urbanucci A, Latonen L, Waltering KK, Sahu B, Jänne OA, et al. Androgen-regulated miR-32 targets BTG2 and is overexpressed in castration-resistant prostate cancer. Oncogene. 2012; 31: 4460-4471. [CrossRef]
- Xiao J, Gong AY, Eischeid AN, Chen D, Deng C, Young CY, et al. miR-141 modulates androgen receptor transcriptional activity in human prostate cancer cells through targeting the small heterodimer partner protein. Prostate. 2012; 72: 1514-1522. [CrossRef]
- Coarfa C, Fiskus W, Eedunuri VK, Rajapakshe K, Foley C, Chew SA, et al. Comprehensive proteomic profiling identifies the androgen receptor axis and other signaling pathways as targets of microRNAs suppressed in metastatic prostate cancer. Oncogene. 2016; 35: 2345-2356. [CrossRef]
- Hagman Z, Haflidadóttir BS, Ceder JA, Larne O, Bjartell A, Lilja H, et al. miR-205 negatively regulates the androgen receptor and is associated with adverse outcome of prostate cancer patients. Br J Cancer. 2013; 108: 1668-1676. [CrossRef]
- Verdoodt B, Neid M, Vogt M, Kuhn V, Liffers ST, Palisaar RJ, et al. MicroRNA-205, a novel regulator of the anti-apoptotic protein Bcl2, is downregulated in prostate cancer. Int J Oncol. 2013; 43: 307-314. [CrossRef]
- Takayama K, Misawa A, Suzuki T, Takagi K, Hayashizaki Y, Fujimura T, et al. TET2 repression by androgen hormone regulates global hydroxymethylation status and prostate cancer progression. Nat Commun 2015; 6: 8219. [CrossRef]
- Shen L, Zhang Y. 5-Hydroxymethylcytosine: generation, fate, and genomic distribution. Curr Opin Cell Biol. 2013; 25: 289-296. [CrossRef]
- Wang Y, Zhang X, Li H, Yu J, Ren X. The role of miRNA-29 family in cancer. Eur J Cell Biol. 2013; 92: 123-128. [CrossRef]
- Langsch S, Baumgartner U, Haemmig S, Schlup C, Schäfer SC, Berezowska S, et al (2016). miR-29b Mediates NF-κB Signaling in KRAS-Induced Non-Small Cell Lung Cancers. Cancer Res. 2016; 76: 4160-4169. [CrossRef]
- Nickerson ML, Im KM, Misner KJ, Tan W, Lou H, Gold B, et al. Somatic alterations contributing to metastasis of a castration-resistant prostate cancer. Hum Mutat. 2013; 34: 1231-1241. [CrossRef]
- Nickerson ML, Das S, Im KM, Turan S, Berndt SI, Li H, et al. TET2 binds the androgen receptor and loss is associated with prostate cancer. Oncogene. 2017; 36: 2172-2183. [CrossRef]
- Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011; 474: 390-394. [CrossRef]
- Hsieh CL, Fei T, Chen Y, Li T, Gao Y, Wang X, et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc Natl Acad Sci U S A. 2014; 111: 7319-7324. [CrossRef]
- Puc J, Kozbial P, Li W, Tan Y, Liu Z, Suter T, et al. Ligand-dependent enhancer activation regulated by topoisomerase-I activity. Cell. 2015; 160: 367-380. [CrossRef]
- Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011; 1: 391-407. [CrossRef]
- Misawa A, Takayama K, Inoue S. Long non-coding RNAs and prostate cancer. Cancer Sci. 2017; 108: 2107-2114. [CrossRef]
- Lanz RB, McKenna NJ, Onate SA, Albrecht U, Wong J, Tsai SY, et al. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell. 1999; 97: 17-27. [CrossRef]
- Lanz RB, Razani B, Goldberg AD, O'Malley BW. Distinct RNA motifs are important for coactivation of steroid hormone receptors by steroid receptor RNA activator (SRA). Proc Natl Acad Sci U S A. 2002; 99: 16081-16086. [CrossRef]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010; 329: 689-693. [CrossRef]
- Yang L, Lin C, Jin C, Yang JC, Tanasa B, Li W, et al. lncRNA-dependent mechanisms of androgen-receptor regulated gene activation programs. Nature. 2013; 500: 598-602. [CrossRef]
- Srikantan V, Zou Z, Petrovics G, Xu L, Augustus M, Davis L, et al. PCGEM1, a prostate-specific gene, is overexpressed in prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 2000; 97: 12216-12221. [CrossRef]
- Petrovics G, Zhang W, Makarem M, Street JP, Connelly R, Sun L, et al. Elevated expression of PCGEM1, a prostate-specific gene with cell growth-promoting function, is associated with high-risk prostate cancer patients. Oncogene. 2004; 23: 605-611. [CrossRef]
- Han Y, Rand KA, Hazelett DJ, Ingles SA, Kittles RA, Strom SS, et al. Prostate cancer susceptibility in men of african ancestry at 8q24. J Natl Cancer Inst. 2016; 108. [CrossRef]
- Misawa A, Takayama K, Urano T, Inoue S et al. Androgen-induced lncRNA SOCS2-AS1 promotes cell growth and inhibits apoptosis in prostate cancer cells. J Biol Chem. 2016; 291: 17861-17880. [CrossRef]
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010; 464: 1071-1076. [CrossRef]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007; 129: 1311-1323. [CrossRef]
- Zhang A, Zhao JC, Kim J, Fong KW, Yang YA, Chakravarti D, et al. LncRNA HOTAIR enhances the androgen-receptor-mediated transcriptional program and drives castration-resistant prostate cancer. Cell Rep. 2015; 13: 209-221. [CrossRef]
- Takayama K, Horie-Inoue K, Katayama S, Suzuki T, Tsutsumi S, Ikeda K, et al. Androgen-responsive long noncoding RNA CTBP1-AS promotes prostate cancer. EMBO J. 2013; 32: 1665-1680. [CrossRef]
- Takayama K, Suzuki T, Fujimura T, Yamada Y, Takahashi S, Homma Y, et al., Dysregulation of spliceosome gene expression in advanced prostate cancer by RNA-binding protein PSF. Proc. Natl. Acad. Sci. U. S. A. 2017; 114: 10461-10466. [CrossRef]
- Lee M, Sadowska A, Bekere I, Ho D, Gully BS, Lu Y, et al. The structure of human SFPQ reveals a coiled-coil mediated polymer essential for functional aggregation in gene regulation. Nucleic Acids Res. 2015; 43: 3826-3840. [CrossRef]
- Prensner JR, Iyer MK, Sahu A, Asangani IA, Cao Q, Patel L, et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat Genet. 2013; 45: 1392-1398. [CrossRef]
- Fei T, Chen Y, Xiao T, Li W, Cato L, Zhang P, et al. Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc Natl Acad Sci U S A. 2017; 114: E5207-E5215. [CrossRef]
