

Uniparental Disomy and Imprinting Disorders
Thomas Eggermann 1, *![]() , Deborah J.G. Mackay 2
, Deborah J.G. Mackay 2![]() , Zeynep Tümer 3, 4
, Zeynep Tümer 3, 4![]()
- Institute of Human Genetics, Medical Faculty, RWTH Aachen University, Aachen, Germany
- Faculty of Medicine, University of Southampton, Southampton SO17 1BJ, UK
- Kennedy Centre, Department of Clinical Genetics, Copenhagen University Hospital, Rigshospitalet, Glostrup, Denmark
- Department of Clinical Medicine, University of Copenhagen, Copenhagen, Denmark
* Correspondence: Thomas Eggermann![]()
Received: June 28, 2018 | Accepted: August 03, 2018 | Published: August 30, 2018
OBM Genetics 2018, Volume 2, Issue 3 doi:10.21926/obm.genet.1803031
Academic Editors: Stéphane Viville and Marcel Mannens
Special Issue: Epigenetic Mechanisms in Health and Disease
Recommended citation: Eggermann T, Mackay DJG, Tümer Z. Uniparental Disomy and Imprinting Disorders. OBM Genetics 2018;2(3):031; doi:10.21926/obm.genet.1803031.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Uniparental disomy (UPD), the inheritance of both homologues of a chromosome from only one parent, has been reported for nearly all human chromosomes. Depending on its mode of formation and time of occurrence, UPD can be present in all cells of an organism, or restricted to some cell lines as a mosaic UPD. Though its general frequency is unknown, it becomes clinically relevant when it produces homozygosity for recessive pathogenic variations or is associated with chromosomal imbalances. UPDs are well-known for their connection to imprinting disorders. Beyond its clinical and diagnostic significance, detection of UPD has value for research in the identification of putative disease mechanisms and genomic regions of interest. Furthermore, detection of UPD in a cluster of similar clinical cases can lead to the definition of new genetic syndromes and imprinted loci, thereby elucidating imprinting regulation and epigenetic mechanisms in general. In this review, we focus on UPDs originating from meiotic and early postzygotic nondisjunction events and their relevance to imprinting disorders.
Keywords
Uniparental disomy; chromosomal non-disjunction; trisomic rescue; monosomic rescue; genomic Imprinting; mosaicism; imprinting disorders; UPD
1. Introduction
Uniparental disomy (UPD), the inheritance of both homologues of a chromosome from only one parent, was first proposed by Eric Engel in 1980 [1], however, another eight years passed before the first case of UPD was reported [2]. Depending on the mode of formation and time of occurrence, UPD can be present in all cells of an organism or restricted to some cell lines as mosaic UPD. In a variety of human cancers, acquired UPD is a common molecular event leading to homozygosity for tumor suppressor genes as well as oncogenes [3].
UPD has pathological significance as it can result in homozygosity for pathogenic gene variations. The term “isozygosity” is used to describe homozygosity of a recessive variant when inherited from the same parent and will be used throughout this review. UPD can also be associated with chromosomal disturbances, UPD can also be associated with chromosomal disturbances, but they are most well-known for their association with imprinting disorders. The identification of UPD in Prader-Willi syndrome (PWS) patients in 1989 [4] demonstrated the importance of UPD in the understanding of genomic imprinting.
In this review, we will focus on UPDs originating from meiotic and early postzygotic nondisjunction events and their relevance for imprinting disorders and their elucidation.
2. Definition and Formation Mechanisms
Unlike normal biparental transmission, UPD occurs when both chromosome homologues are inherited from a single parent. An individual with both homologous chromosomes (genetically different) from the same parent is said to have uniparental heterodisomy (UPhD), whereas deriving homologous (genetically identical) chromosomes from the same parent is referred to as uniparental isodisomy (UPiD). Heterodisomy typically occurs as a result of meiosis I nondisjunction, while isodisomy is typically due to a meiosis II nondisjunction or a mitotic error. Mixed forms of UPiD/UPhD or UPhD/UPiD are also possible (Figure 1a). Furthermore, UPDs can be present with or without chromosomal aberrations, which include mosaic trisomy, translocations, and isochromosomes [5]. UPDs can involve a whole chromosome or can be segmental, where only part of a chromosome is inherited from the same parent. Additionally, the whole chromosome complement (whole genome) can be derived from one parent (uniparental diploidy), either from the mother or from the father. The term “genome-wide UPD” is commonly used in literature, but we suggest using the term “uniparental diploidy” instead.



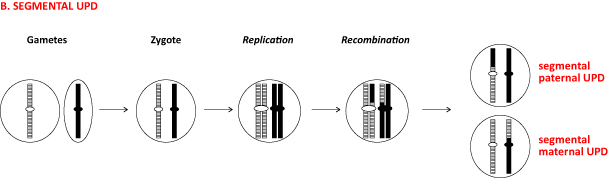

Figure 1 Example mechanisms of UPD formation. (a) Four scenarios leading to whole chromosome UPDs. In the figure, only monosomy rescue for maternal UPD is shown; paternal UPD is caused by monosomy rescue following fertilization between nullisomic oocyte and normal sperm. Paternal chromosomes are shown in black; maternal homologous chromosomes are striped. Small horizontal ellipses depict centromeres. UPhD, uniparental heterodisomy; UPiD, uniparental isodisomy. (b) Mechanism leading to segmental UPD. (c) UPD mechanisms in the offspring of Robertsonian translocation carriers. The mother is carrier of t (14; 22) translocation, while the father has normal karyotype. Chromosome 22 is shown in black (paternal) or gray (maternal). Maternal homologue chromosomes 14 are shown in red and pink, and paternal chromosomes 14 in dark and light blue.
2.1. Mechanisms of Whole Chromosome UPD Formation
Whole chromosome UPDs occur via four main mechanisms (Figure 1a). The first three mechanisms originate in meiosis and are a corrective response to aneuploidy. Human oocytes are markedly prone to aneuploidy, particularly with increasing maternal age [6] and in association with defects in meiotic crossover [7]; premature separation is suggested as the major segregation error [8]. Uniparental disomy represents a potential mechanism for aneuploidy rescue and likewise shows increasing frequency with maternal age [9,10].
Trisomic rescue. Trisomic rescue happens when a meiotic nondisjunction occurs in one of the conceiving gametes resulting in disomy and a subsequent trisomic conceptus after fertilization. The trisomy is corrected by non-random elimination of the extra chromosome (trisomic rescue); if the remaining chromosomes are from the same parent, this will result in uniparental disomy (UPD). Depending on whether the nondisjunction occurred during meiosis I or II, the UPD can be heterodisomic (UPhD) or isodisomic (UPiD), respectively (if we assume recombination between sister chromatids has not occurred). Mixed UPhD/UPiD or UPiD/UPhD (the centromere are within the heterodisomic region, and the centromere are within the isodisomic region, respectively) are caused by recombination between the sister chromatids of a pair of the homologous chromosomes of the disomic conceiving gamete (Figure 1a). UPhD/UPiD, with heterodisomic markers around the centromere and isodisomic markers near the telomere, indicates a meiosis I error, whereas UPiD/UPhD with isodisomic markers around the centromere and heterodisomic markers near the telomere indicates a meiosis II error (Figure 1a). If the trisomic rescue occurs in a later postzygotic stage, this will result in a mosaic UPD case [11].
Monosomic rescue. In this model, nondisjunction during meiosis I in one of the conceiving gametes results in nullisomy and in a monosomic conceptus after fertilization. Monosomic rescue takes place in the early postzygotic stage by replication of the monosomic chromosome, leading to UPiD.
Postfertilization (mitotic) error. In this case the conceptus is disomic. A mitotic error can lead to a monosomy or trisomy that can be corrected through monosomic or trisomic rescue, respectively, and may lead to isodisomic UPD in both scenarios.
Gamete complementation. This mechanism requires simultaneous occurrence of meiotic disjunction in both gametes, where one is nullisomic and the other is disomic, leading to UPhD in the conceptus. If a recombination had occurred between the sister chromatids of a pair of the homologous chromosomes of the disomic conceiving gamete, this will lead to mixed UPiD/UPhD (centromere is within the isodisomic region) or UPhD/UPiD (centromere is within the heterodisomic region) (Figure 1a).
2.2. Mechanisms of Segmental UPD Formation
Segmental UPD is one of the main molecular alterations observed in Beckwith-Wiedemann syndrome [12]. It is suggested to result from a postzygotic mitotic cross-over between the paternal and maternal homologue chromosomes and in principle can only be a segmental isodisomy (Figure 1b). An alternative mechanism is trisomic rescue as described above, where a mitotic recombination between paternal and maternal homologue chromosomes occurs before the “normal” extra chromosome is discarded from the cell. In this mechanism, segmental UPD can be both isodisomic and heterodisomic. Segmental UPD can also be mosaic [12].
2.3. Mechanisms for Uniparental Diploidy
Only mosaic uniparental diploidy conceptuses are viable and most of these are of paternal origin [13,14]; only one single case of maternal uniparental diploidy has been described in literature [15]. Four different mechanisms have been suggested for mosaic uniparental diploidy occurrence and were described in detail by Kalish et al. [13].
2.4. UPDs Accompanied by Chromosome Aberrations
UPD cases may arise due to translocations [5]. Different scenarios have been proposed for the formation of UPD in the offspring of the carriers of balanced translocations, including Robertsonian translocations (Figure 1c) [5]. The karyotypes of these offspring may be balanced or unbalanced. In one of these scenarios, the chromosome aberration of the offspring with UPiD is an isochromosome (homologous Robertsonian translocation), which likely occurs from a monosomic rescue. Notably, upd(14) and upd(15) are the most frequently reported UPDs associated with Robertsonian translocations [5], but this might be due to the fact that the majority of Robertsonian translocations affect chromosome 14 and that clinical pictures are only associated with UPDs of these chromosomes; UPDs of chromosomes 13, 21, and 22 have not yet been reported as clinically relevant (Table 1).
UPDs can also be accompanied by small supernumerary marker chromosomes and the formation mechanisms are explained in detail by Liehr [5].
Table 1 Congenital imprinting disorders associated with UPDs
|
In ten of the 13 UPDs, the role of disturbed imprinting as the cause for the associated phenotypes is evident, but in three it is in discussion (in italics).
3. Epidemiology
Maternal and paternal UPDs have been described for all human chromosomes except 19 and Y, and more than 3,600 cases with UPDs in developmental disorders are documented [16]. In the majority of UPDs, no specific phenotypes are associated with the aberrant constitution, but an association with clinical pictures has been shown for ten UPDs (Table 1), which are distinguished as imprinting disorders.
The total frequency of all UPDs (including whole chromosome and segmental UPDs) is unknown, because (a) there have been no systematic studies to determine its prevalence, (b) UPDs without clinical consequences are not identified, and (c) UPD mosaicism cannot readily be detected using standard diagnostic algorithms. In fact, the majority of UPDs have been identified adventitiously in individuals with clinical features or indications for UPD testing (e.g. imprinting disorders and specific chromosomal variants).
Through extrapolation of UPD frequency of chromosome 15, Robinson suggested the general incidence of UPD as 1:3,500 [6]. Its incidence appears increased in patients exhibiting intellectual disability, developmental delay, dysmorphisms, and congenital anomalies. In 2010, Conlin et al. [17] reported on eight patients with (segmental) UPD among 2019 patients (0.39%). A similar ratio was determined in a larger cohort of 14,574 patients by Wang et al. [18], who identified a UPiD in 0.13% (n = 19), and a mixed UPhD/UPiD or segmental UPD in 0.17% (n = 25).
More recently, further estimates have been based on array analyses. Using chromosome microarray data from 173 trios, Sasaki et al. [19] estimated a rate of approximately 0.6% for segmental UPDs. In a cohort of 14,574 patients with a broad range of clinical indications, Wang et al. [18] identified 25 cases with significant regions of homozygosity of a single chromosome. In five of those patients, mixed UPhD/UPiD could be confirmed, and in 2 patients a segmental UPD was identified. In the remaining cases UPD was suspected. Thus, an incidence of less than 1% in patients with clinical features can be assumed. However, this data is limited by a defined restriction of a region of homozygosity of ≥5 Mb [18]; therefore, segmental UPiDs/UPhDs with a size of less than 5 Mb escape detection.
Chromosome-specific frequencies of UPDs are difficult to estimate as there are strong ascertainment biases. An example is upd(15)mat: it is overwhelmingly overrepresented in reports on UPD because upd(15)mat accounts for 70-75% of Prader-Willi syndrome (PWS) occurences, a condition that is well-known in pediatrics and therefore frequently tested. In contrast, upd(15)pat is less frequently documented as it accounts for only 1-2% of the mutation spectrum in Angelman syndrome (AS), an imprinting disorder with a similar prevalence as PWS. This ascertainment bias is obvious in the studies of Conlin et al. and Wang et al. [17,18], where a considerable number of UPDs were upd(15)mat. However, two further UPD constitutions, upd(7)mat and upd(14)mat, are frequently published although the associated disorders are rare. Many groups working in the field of imprinting disorders have recently focused on these specific alterations and have screened for specific clinical cohorts.
Aside from imprinting disorders, UPDs are also associated with chromosomes affected by trisomy. For example, trisomy 16 (with maternal origin of the supernumerary chromosome) is one of the most common aneuploidies detected during human prenatal development; while it persists in growing placenta, it is incompatible with fetal development and therefore is reduced by trisomy rescue to give a relatively high frequency of upd(16)mat in liveborn offspring [14].
In summary, the current observed frequency of UPDs is determined by the observance of clinical consequences and associations with other chromosomal aberrations, but is likely not accounting for UPDs that do not have clinical presentations. It is likely to hypothesize that the actual frequency of UPDs is higher than the current observed frequency.
4. Risk Factors for UPD Formation Following Meiotic and Early Postzygotic Nondisjunction
Individuals who are at risk to carry UPDs are (a) children born to older mothers, (b) children from pregnancies with chromosomal aneuploidy mosaicism in the extraembryonic tissues, and (c) carriers of (familial) structural variants or their offspring (i.e. Robertsonian translocations or marker chromosomes).
The association of UPD with advanced maternal age can be explained by the increased risk of trisomy formation of older mothers and the subsequent trisomic rescue, which is the major mode of UhPD formation (Figure 1). This association only applies to maternal UPhD, however it can be assumed that increased maternal age is the major risk factor for UPD formation. Its relevance has already been proven for several UPDs (e.g. upd(20)mat and upd(16)mat [20,21]). In contrast, paternal UPDs and UPiDs should not be influenced by maternal age in theory, and this has been confirmed for upd(7)mat where the maternal age in the UPhD subcohort is higher than in the UPiD group [22].
Trisomy rescue also explains the occurrence of UPD in pregnancies with chromosomal aberrations detected during prenatal development. Trisomy rescue can typically be detected by prenatal identification of a trisomy in a chorionic villous sample followed by identification of a disomy in a subsequent amniocentesis. This mechanism was first documented in 1992 for chromosome 15, with a with a 47, +15 karyotype at chorionic villous sampling, a normal karyotype in amniocentesis, and a upd (15) mat in the newborn [23,24]. It is therefore recommended to test fetuses of pregnancies with trisomic cells in chorionic villous sampling for UPD, at least for those with a clinical relevance [25].
Carriers of (balanced) structural chromosomal variants or their offspring have an increased risk for UPD (see above). In this category, Robertsonian translocations affecting acrocentric chromosomes are the most frequent structural variants in humans (incidence in the general population: 0.1%). Robertsonian translocations involving chromosomes 14 and 15 lead to increased risk of four imprinting disorders in affected families. These types of translocations gave the first clues towards the identification of the imprinted region in 15q11-q13 and the associated imprinting disorders PWS and AS [26]. It also subsequently allowed the identification of the chromosome 14-associated imprinting disorders [27,28]. However, translocations other than the acrocentric chromosomes should be regarded as risk factors for UPD formation, as documented by several case reports [29]. In families with a known translocation or a prenatal detection of a variant, UPD testing is suggested if there is evidence for involvement of a chromosome containing imprinted genes, i.e. chromosomes 6, 7, 11, 14, 15,and 20 [25]. Another group of structural variants which might be associated with UPD are small supernumerary marker chromosomes, however, this combination has rarely been reported [30,31]. Regardless, UPD testing should be considered in cases where clinical features are compatible with an imprinting disorder, or when chromosomes containing imprinted genes are involved.
The identification of structural variants underlying UPD is essential to give accurate genetic counselling of recurrence risk. In families with structural chromosomal variants, there may be elevated risk not only for UPD, but also for unbalanced chromosomal rearrangements.
5. UPDs and Their (Patho) Physiological Significance
Though UPDs themselves do not alter the karyotype or DNA sequence, they can be associated with an aberrant phenotype due to the three following mechanisms.
5.1. UPDs and Genomic Imprinting
Genomic imprinting is the restriction of gene expression potential by parent of origin [32]. Approximately 40 human genomic regions evade epigenetic reprogramming in the early embryo, and thus faithfully maintain the epigenetic programming of the egg or sperm. As a result, at key developmental times and places the expression of imprinted genes is functionally hemizygous. Disturbance of this epigenetically-balanced gene expression at certain loci gives rise to imprinting disorders (Figure 2).
In a UPD affecting an imprinted locus, methylation is faithfully maintained. As both alleles are from the same parent, no expression of the imprinted genes within this locus occurs, leading to disturbance of the overall balance of methylation and therefore gene expression.
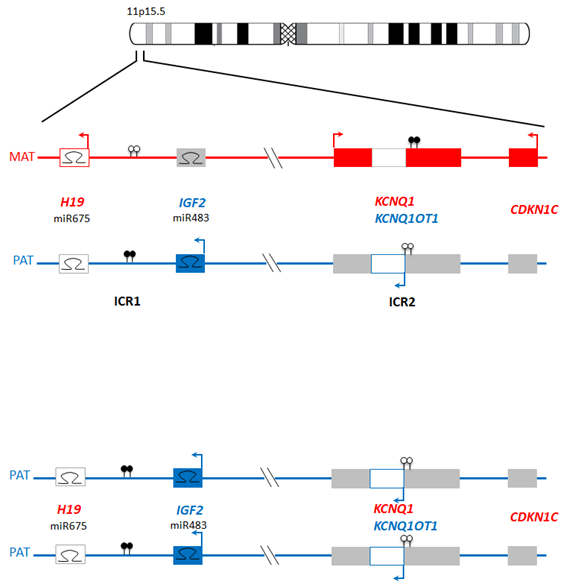
Figure 2 Example of imprinting regulation utilizing the imprinted regions on chr11p15. The upper panel represents normal biparental inheritance at the closely apposed imprinted loci on chr11p15. The lower panel represents paternal UPD, where the balance of imprinted gene expression is disturbed via downregulation of H19 and CDKN1C and upregulation of IGF2. Maternally and paternally expressed genes are shown with red and blue horizontal blocks, respectively. Filled lollipops, methylated CpG sites; empty lollipops, unmethylated CpG sites. Arrows show direction of transcription, and Ω symbols represent micro-RNA binding sites.
5.2. UPDs and Chromosomal Aberrations/Structural Variants
The phenotype of UPD carriers can be influenced by a co-occurring trisomy or structural variant. In this situation, the UPD is regarded as a biomarker for the chromosomal alteration (e.g. upd(6)mat and upd(16)mat) [20,33] rather than as the cause of the aberrant phenotype. Moreover, the combination of both chromosomal alterations and UPDs can cause heterogeneous clinical presentations, obscuring clinical diagnosis or mimicking known disorders.
5.3. UPDs as Cause of Unexpected Homozygosity for Recessive Mutations - Isozygosity
Apart from the UPDs of chromosomes harboring imprinted genes (Table 1), maternal and paternal UPDs of other chromosomes have been identified by chance, either as coincidental findings by genotyping assays (SNP arrays and NGS) in research or diagnostic contexts (e.g. GWAS in research context and molecular karyotyping in diagnostics), or in patients with unexpected isozygosity by linkage and mutation analysis. In these families, only one of the parents is a heterozygous carrier, and the child is homozygous due to UPiD. In fact, this was the case in the first proven patient with UPD, suffering from cystic fibrosis due to maternal UPiD of chromosome 7 and homozygosity for a CFTR variant [2]. UPiD can be restricted to the region harboring the gene affected by the pathogenic variant or affect the whole chromosome. In many cases, the UPiD has been identified after discovery of the homozygous pathogenetic variant, but identification of UPiD can also be the first step in the detection of a candidate gene and the pathogenic mutation [18].
6. UPDs and Their Clinical Significance
According to the aforementioned pathomechanisms, UPDs can affect phenotype by the homozygosity of a recessive allele or an associated chromosomal disturbance. In the latter case, UPD can be regarded as one molecular type out of a variety of molecular pathomechanisms on a genomic level that result in the gene and mutation-specific phenotype.
Because of their association with imprinting disorders, UPDs have clinical signficance (Table 2).
Imprinting disorders are a group of rare human congenital diseases with prevalences less than 1:10,000 among newborns. These disorders are caused by changes in both gene sequences (“genetic variations,” which include UPDs) and gene regulation (“epigenetic variations”), affecting parentally-imprinted loci. To date, twelve clinically-recognized disorders have been defined and characterized by common underlying molecular mechanisms and overlapping phenotypes; in ten of these UPDs have been identified [32]. Clinical features primarily appear prenatally or in early childhood, and nearly all of them have a severe impact on health including abnormal growth, tumor development, disturbed metabolism, neurodevelopmental delay, and precocious puberty. They include well-known congenital disorders like PWS, AS, Beckwith-Wiedemann, and Silver-Russell syndrome, as well as rare disorders that are lesser-known such as transient neonatal diabetes mellitus; pseudohypoparathyroidism; or the recently described Temple, Kagami-Ogata, Schaaf-Yang, and Mulchandani-Bhoj-Conlin syndromes. Recently, upd(6)mat, upd(16)mat, and upd(7)pat have been suggested to be associated with imprinting disorders [20,33,34]. However, the influence of trisomy 6 or 16 mosaicism on the phenotype has been suggested for upd(6)mat and upd(16)mat, based on the genetic findings and the clinical heterogeneity of the carriers.
Due to the clinical heterogeneity of imprinting disorders and their ambiguity to physicians, patients often experience a delay in diagnosis, or remain without diagnosis. As a result, the prevalence of imprinting disorders among newborns are currently unknown and can be only roughly estimated. The frequent presence of non-specific features and the phenotypic overlaps between the different disorders as well as variance in diagnoses challenges the accurate diagnosis of imprinting disorders (Table 2). In addition, the classification of imprinting disorders is becoming increasingly complex, causing further complexity in diagnosis and leading to uncertainty for patients and families, as well as healthcare providers.
Table 2 Summary of the most frequent and overlapping features in imprinting disorders

(- not present; + present; NR not reported but possible; ? unknown or reported in rare cases, therefore making an association questionable)
7. UPD as a Tool for Research
Beyond its diagnostic significance, detection of UPD has been valuable for research: (a) its detection during genome-wide genetic diagnosis can suggest a new disease mechanism and a genomic region of interest, (b) detection in a cluster of similar clinical cases can lead to defining new genetic syndromes, and (c) aiding in the identification of imprinted genes.
7.1. Adventitious Detection Suggesting Disease Mechanisms and Their loci
Identification of UPDs has been a significant by-product of genetic diagnosis from its inception. For example, the first association of UPD with Silver-Russel syndrome occured during discovery of recessive cystic fibrosis [2,35]; the diagnosis of cystic fibrosis continues to provide clarification of the gene(s) involved with SRS on chr7 [36].
Additionally, the current large-scale genomic medicine initiatives offer a very welcome opportunity to detect UPD [37]. Moreover, researchers and diagnosticians across the world ought to continue to share these rare diagnoses, in order to identify clusters of similar individuals (see below). In some cases, UPD may be coincidental to clinical presentation, or may be a manifestation of another disease mechanism. For instance, cases in literature describe UPD in association with clinical presentations [34,37]; time will tell whether these descriptions will develop into new clinical UPD disorders, represent coincidental findings, or reveal recessive disorders.
7.2. Clustering of Cases with Similar Clinical Presentations and Shared UPD May Lead to The Recognition of New Clinical Syndromes
An example of a specific syndrome that originated from a cluster of cases with similar clinical presentations is the isolated syndromic presentation of upd(14)mat. Although it was first described in 1991 [28], it was recently recognized as a distinct syndrome and named Temple syndrome (TS14) [38,39], and has a degree of phenotypic overlap with both PWS and SRS [40]. Defining the clinical parameters of TS14 led to refinement of its genetic etiology [41], and it is now routinely identified in association with CNVs, UPD, and epimutations (i.e. aberrant methylation marks without obvious alteration of the DNA sequence) of the imprinted gene cluster on chr14q32. A similar process of clinical clustering and syndrome definition is in progress with upd(20)mat [21,42], which ought to refine its genetic etiology.
7.3. With the Advent of Genome-wide DNA Methylation Analysis, DNA from Individuals with UPD has Been Used to Identify Imprinted Genes
Comparative analysis, such as genome-wide DNA methylation analysis, can either use DNA from numerous individuals with UPD of different chromosomes[43], uniparental diploidy [44], or nonviable imprinting errors such as hydatidiform moles and ovarian teratomas [45]. These analyses have yielded what can reasonably be claimed as a comprehensive list of germline-imprinted genes (i.e. genes where the imprinting marks are established in the gametes), which has been invaluable in understanding the mechanisms and pathologies of imprinting. Notably, placental imprinting and its pathologies are relatively less understood [46].
8. Methods to Identify UPD
The appropriate tools to identify UPDs are DNA-based polymorphic markers (Table 3). In contrast, tests visualizing chromosomes (including conventional karyotyping), fluorescence in situ hybridization (FISH), and chromosome microarrays (SNP-arrays or array CGH) for detection of copy number variants (CNVs), are not suitable to detect UPDs; these can only suggest chromosomal constitutions prone to UPD formation, like trisomy mosaicism or structural variants which might result in UPD (e.g. t(14;14) [25]).
In principle, UPDs can be identified by all techniques that can detect the genotyping of polymorphic markers (Figure 3).
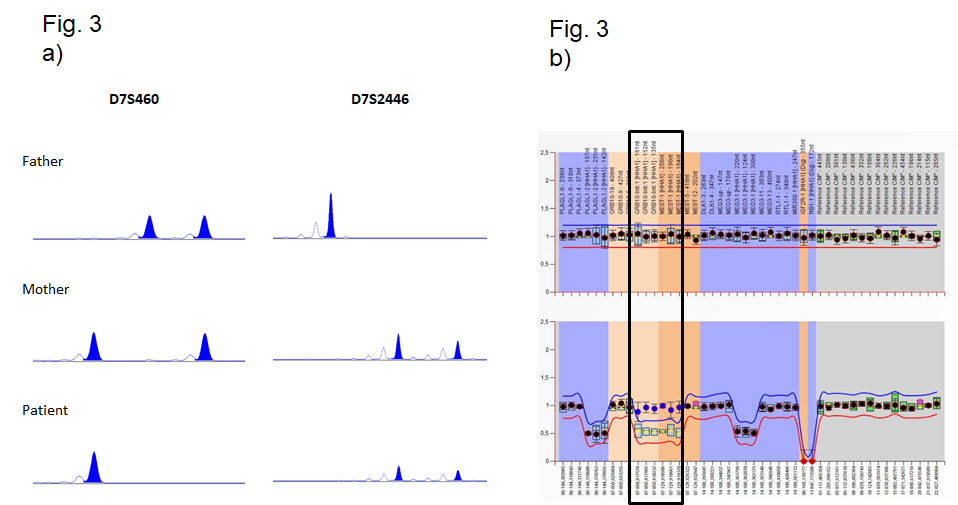
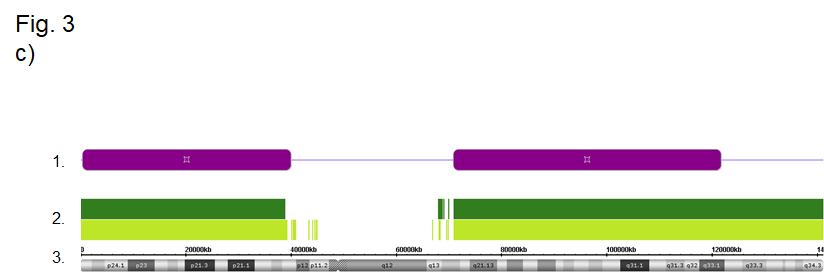
Figure 3 Identification of UPD by molecular genetic testing. (a, b) Identification of upd(7)mat by microsatellite typing and methylation-specific MLPA: (a) Typing results of the marker D7S460 reveals a UPiD, whereas typing results for D7S2446 correspond to a UPhD. Please note that the typing result of D7S460 does not allow the discrimination between UPiD and a deletion of the paternal allele. (b) By MS-MLPA, a deletion of the imprinted genes GRB10 (7p13) and MEST (7q32) could be excluded by copy number analysis (upper row), but methylation analysis (lower row) does not discriminate between upd(7)mat and isolated hypermethylation (epimutation). (c) Identification of a UPiD of chromosome 9 by SNP array analysis (Cytoscan® HD, Affymetrix, Santa Clara/CA, USA): (1) large stretches of loss of heterozygosity indicate a uniparental isodisomy for these regions. (2) Distributi on of SNP (light green) and oligo probes (dark green). (3) Physical map of chromosome 9.
Table 3 Methods applicable for identification and proof of UPDs and other molecular changes in imprinting disorders.
These can be highly polymorphic markers like microsatellites (also referred to as short tandem repeats) or single nucleotide variants. The classical tool utilized to identify UPDs is microsatellite typing, and it has been determined that two informative markers (meaning that both parents are heterozygous for different alleles) are sufficient to confirm a UPD [25]. Microsatellite typing is based on PCR-based amplification of highly polymorphic repeats (e.g. CA repeats or trinucleotide repeats), followed by high-resolution gel electrophoresis. It includes parallel analysis of DNA samples of the patient and at least one parent. In the case of a positive testing result, both parental samples should be included, and paternity should be confirmed for maternal UPD. Microsatellite typing is well-established in routine diagnostic labs as it requires basic molecular laboratory equipment and is therefore fast and cheap. It is currently utilized as the major test in the diagnosis of UPD, however high-throughput genotyping techniques (such as SNP array analysis) are growing in significance. The advantage of this test is that thousands of SNP markers on one chromosome are analyzed, thereby even small segmental UPDs as well as uniparental diploidy are detectable. However, the identification of UPhD needs analyses of parental samples and specific software tools [47], which is much more expensive than microsatellite analysis.
In the case of specific chromosomes that contain imprinted loci, methylation-specific (MS) tests can provide indirect evidence for a UPD. As illustrated in Figure 3, MS multiplex ligation-dependent probe amplification (MS-MLPA) is a suitable assay to identify disturbed methylation patterns, with the advantage that it addresses copy number variations and methylation marks in one assay, and can target more than 40 loci. However, it should be noted that MS-MLPA, like other MS assays (Table 3), can identify copy number variations, epimutations, and UPD, but might not discriminate epimutations from UPD.
Nevertheless, molecular diagnostic testing is currently being revolutionized by the implementation of NGS-based assays, which also affects UPD testing. In the future, the choice of which method to apply will depend on the indication: in the case of targeted UPD testing, e.g. in families with Robertsonian translocations, short tandem repeat typing might be sufficient, whereas in nonspecific phenotypes, array or NGS testing might be indicated.
Acknowledgement
We thank Jette Bune Rasmussen for assistance in drawing Figure 1
Author Contributions
All authors contributed to the preparation of the paper and extensively discussed the contents and structures.
Funding Source
The authors have been members of the COST Action BM1208 (European Network of Congenital Imprinting disorders). TE is supported by the Deutsche Forschungsgemeinschaft (DFG, EG110/15-1).
Competing Interests
The authors have declared that no competing interests exist.
References
- Engel E. A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am J Med Genet. 1980; 6: 137–143. [CrossRef] [Google scholar] [PubMed]
- Spence JE, Perciaccante RG, Greig GM, Willard HF, Ledbetter DH, Hejtmancik JF, et al. Uniparental disomy as a mechanism for human genetic disease. Am J Hum Genet. 1988; 42: 217-226. [Google scholar]
- Makishima H, Maciejewski JP. Pathogenesis and Consequences of Uniparental Disomy in Cancer. Clin Cancer Res. 2011; 17: 3913-3923. [CrossRef] [Google scholar] [PubMed]
- Nicholls RD, Knoll JHM, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in non-deletion Prader-Willi syndrome. Nature. 1989; 342: 281-285. [CrossRef] [Google scholar] [PubMed]
- Liehr T. Uniparental Disomy (UPD) in Clinical Genetics. Springer Berlin Heidelberg. 2014. [CrossRef] [Google scholar]
- Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays. 2000; 22: 452-459. [CrossRef] [Google scholar]
- Wang S, Hassold T, Hunt P, White MA, Zickler D, Kleckner N, et al. Inefficient Crossover Maturation Underlies Elevated Aneuploidy in Human Female Meiosis. Cell. 2017; 168: 977-989. [CrossRef] [Google scholar] [PubMed]
- Yan Y, Lane SIR, Jones KT. Premature dyad separation in meiosis II is the major segregation error with maternal age in mouse oocytes. Development. 2014; 141: 199-208. [CrossRef] [Google scholar] [PubMed]
- Robinson WP, Lorda-Sanchez I, Malcolm S, Langlois S, Schuffenhauer S, Knoblauch H, et al. Increased parental ages and uniparental disomy 15: a paternal age effect? Eur J Hum Genet. 1993; 1: 280-286. [CrossRef] [Google scholar] [PubMed]
- Kotzot D. Advanced parental age in maternal uniparental disomy (UPD): implications for the mechanism of formation. Eur J Hum Genet. 2004; 12: 343-346. [CrossRef] [Google scholar] [PubMed]
- Gardner RJM, Sutherland GR, Shaffer LG. Chromosome Abnormalities and Genetic Counseling (4 ed.). Oxford University Press. 2004; 250-250 p. [Google scholar] [PubMed]
- Keren B, Chantot-Bastaraud S, Brioude F, Mach C, Fonteneau E, Azzi S, et al. SNP arrays in Beckwith-Wiedemann syndrome: an improved diagnostic strategy. Eur J Med Genet. 2013; 56: 546-550. [CrossRef] [Google scholar] [PubMed]
- Kalish JM, Conlin LK, Bhatti TR, Dubbs HA, Harris MC, Izumi K, et al. Clinical features of three girls with mosaic genome-wide paternal uniparental isodisomy. Am J Med Genet A. 2013; 161: 1929-1939. [CrossRef] [Google scholar] [PubMed]
- Borgulová I, Soldatova I, Putzová M, Malíková M, Neupauerová J, Marková SP, et al. Genome-wide uniparental diploidy of all paternal chromosomes in an 11-year-old girl with deafness and without malignancy. J Hum Genet. 2018; 63: 803-810. [CrossRef] [Google scholar] [PubMed]
- Yamazawa K, Nakabayashi K, Kagami M, Sato T, Saitoh S, Horikawa R, et al. Parthenogenetic chimaerism/mosaicism with a Silver-Russell syndrome-like phenotype. J Med Genet. 2010; 47: 782-785. [CrossRef] [Google scholar] [PubMed]
- Liehr T. Cytogenetic contribution to uniparental disomy (UPD). Mol Cytogenet. 2010; 3: 8-8. [CrossRef] [Google scholar] [PubMed]
- Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, et al. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet. 2010; 19: 1263-1275. [CrossRef] [Google scholar] [PubMed]
- Wang JC, Ross L, Mahon LW, Owen R, Hemmat M, Wang BT, et al. Regions of homozygosity identified by oligonucleotide SNP arrays: evaluating the incidence and clinical utility. Eur J Hum Genet. 2015; 23: 663-671. [CrossRef] [Google scholar] [PubMed]
- Sasaki, Kensaku, Mishima, Hiroyuki, Yoshiura, Koh-ichiro, et al. Uniparental disomy analysis in trios using genome-wide SNP array and;whole-genome sequencing data imply segmental uniparental isodisomy in;general populations. Gene. 2013; 512: 267-274. [CrossRef] [Google scholar] [PubMed]
- Scheuvens R, Begemann M, Soellner L, Meschede D, Raabe‐Meyer G, Elbracht M, et al. Maternal uniparental disomy of chromosome 16 (upd(16)mat): Clinical features are rather caused by (hidden) trisomy 16 mosaicism than by upd(16)mat itself. Clin Genet. 2016; 92: 45-51. [CrossRef] [Google scholar] [PubMed]
- Mulchandani S, Bhoj EJ, Luo M, Powellhamilton N, Jenny K, Gripp KW, et al. Maternal uniparental disomy of chromosome 20: a novel imprinting disorder of growth failure. Genet Med. 2015; 18: 309-315. [CrossRef] [Google scholar] [PubMed]
- Chantot-Bastaraud S, Stratmann S, Brioude F, Begemann M, Elbracht M, Graul-Neumann L, et al. Formation of upd(7)mat by trisomic rescue: SNP array typing provides new insights in chromosomal nondisjunction. Mol Cytogenet. 2017; 10: 28. [CrossRef] [Google scholar] [PubMed]
- Cassidy SB, Lai LW, Erickson RP, Magnuson L, Thomas E, Gendron R, et al. Trisomy 15 with loss of the paternal 15 as a cause of Prader-Willi syndrome due to maternal disomy. Am J Hum Genet. 1992; 51: 701-708. [Google scholar]
- Purvis-Smith SG, Saville T, Manass S, Yip MY, Lam-Po-Tang PR, Duffy B, et al. Uniparental disomy 15 resulting from “correction” of an initial trisomy 15. Am J Hum Genet. 1992; 50: 1348-1350. [Google scholar]
- Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med. 2001; 3: 139-148. [CrossRef] [Google scholar] [PubMed]
- Smith A, Noel M. A girl with the Prader-Willi Syndrome and Robertsonian translocation 45,XX,t(14;15)(p11;q11) which was present in three normal family members. Hum Genet. 1980; 55: 271-273. [CrossRef] [Google scholar] [PubMed]
- Wang JC, Passage MB, Yen PH, Shapiro LJ, Mohandas TK. Uniparental heterodisomy for chromosome 14 in a phenotypically abnormal familial balanced 13/14 Robertsonian translocation carrier. Am J Hum Genet. 1991; 48: 1069-1074. [Google scholar]
- Coviello DA, Panucci E, Mantero MM, Perfumo C, Guelfi M, Borrone C, et al. Maternal uniparental disomy for chromosome 14. Acta Genet Med Gemellol. 1991; 28: 169-172. [Google scholar]
- Behnecke A, Hinderhofer K, Jauch A, Janssen JW, Moog U. Silver-Russell syndrome due to maternal uniparental disomy 7 and a familial reciprocal translocation t(7;13). Clin Genet. 2012; 82: 494-498. [CrossRef] [Google scholar] [PubMed]
- Liehr T, Ewers E, Hamid AB, Kosyakova N, Voigt M, Weise A, et al. Small supernumerary marker chromosomes and uniparental disomy have a story to tell. J Histochem Cytochem. 2011; 59: 842-848. [CrossRef] [Google scholar] [PubMed]
- Liehr T, Mrasek K, Kosyakova N, Ogilvie CM, Vermeesch J, Trifonov V, et al. Small supernumerary marker chromosomes (sSMC) in humans; are there B chromosomes hidden among them. Mol Cytogenet. 2008; 1: 12-12. [CrossRef] [Google scholar] [PubMed]
- Djg M. Human imprinting disorders: Principles, practice, problems and progress. Eur J Med Genet. 2017; 60: 618-626. [CrossRef] [Google scholar] [PubMed]
- Eggermann T, Oehl‐Jaschkowitz B, Dicks S, Thomas W, Kanber D, Albrecht B, et al. The maternal uniparental disomy of chromosome 6(upd(6)mat) “phenotype”: result of placental trisomy6mosaicism? Mol Genet Genomic Med. 2017; 5: 668-677. [CrossRef] [Google scholar] [PubMed]
- Nakamura A, Muroya K, Ogata-Kawata H, Nakabayashi K, Matsubara K, Ogata T, et al. A case of paternal uniparental isodisomy for chromosome 7 associated with overgrowth. J Med Genet. 2018; jmedgenet-2017-104986. [Google scholar]
- Voss R. Isodisomy of chromosome 7 in a patient with cystic fibrosis: could uniparental disomy be common in humans? Am J Hum Genet. 1989; 45: 373-380. [CrossRef] [Google scholar]
- Gerbrands LC, Haarman EG, Hankel MA, Finken MJJ. Cystic fibrosis and Silver–Russell syndrome due to a partial maternal isodisomy of chromosome 7. Clinical Case Reports. 2017; 5: 1697-1700. [CrossRef] [Google scholar] [PubMed]
- King DA, Fitzgerald TW, Miller R, Canham N, Claytonsmith J, Johnson D, et al. A novel method for detecting uniparental disomy from trio genotypes identifies a significant excess in children with developmental disorders. Genome Res. 2014; 24: 673-687. [CrossRef] [Google scholar] [PubMed]
- Temple IK, Shrubb V, Lever M, Bullman H, Mackay DJG. Findings that shed new light on the possible pathogenesis of a disease or an adverse effect: Isolated imprinting mutation of the DLK1/GTL2 locus associated with a clinical presentation of maternal uniparental disomy of chromosome 14. Bmj Case Rep. 2009; 2009. [Google scholar]
- Ioannides Y, Lokulosodipe K, Mackay DJG, Davies JH, Temple IK. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J Med Genet. 2014; 51: 495-501. [CrossRef] [Google scholar] [PubMed]
- Kagami M, Mizuno S, Matsubara K, Nakabayashi K, Sano S, Fuke T, et al. Epimutations of the IG-DMR and the MEG3-DMR at the 14q32.2 imprinted region in two patients with Silver|[ndash]|Russell Syndrome-compatible phenotype. Eur J Hum Genet. 2015; 23: 1062-1067. [CrossRef] [Google scholar] [PubMed]
- Kagami M, Sekita Y, Nishimura G, Irie M, Kato F, Okada M, et al. Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat Genet. 2008; 40: 237-242. [CrossRef] [Google scholar] [PubMed]
- Kawashima S, Nakamura A, Inoue T, Matsubara K, Horikawa R, Wakui K, et al. Maternal Uniparental Disomy for Chromosome 20: Physical and Endocrinological Characteristics of Five Patients. J Clin Endocrinol Metab. 2018; 103: 2083. [CrossRef] [Google scholar] [PubMed]
- Joshi RS, Garg P, Zaitlen N, Lappalainen T, Watson CT, Azam N, et al. DNA Methylation Profiling of Uniparental Disomy Subjects Provides a Map of Parental Epigenetic Bias in the Human Genome. Am J Hum Genet. 2016; 99: 555-566. [CrossRef] [Google scholar] [PubMed]
- Nakabayashi K, Trujillo AM, Tayama C, Camprubi C, Yoshida W, Lapunzina P, et al. Methylation screening of reciprocal genome-wide UPDs identifies novel human-specific imprinted genes. Hum Mol Genet. 2011; 20: 3188-3197. [CrossRef] [Google scholar] [PubMed]
- Court F, Tayama C, Romanelli V, Martin-Trujillo A, Iglesias-Platas I, Okamura K, et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014; 24: 554-569. [CrossRef] [Google scholar] [PubMed]
- Sanchezdelgado M, Court F, Vidal E, Medrano J, Monteagudosánchez A, Martintrujillo A, et al. Human Oocyte-Derived Methylation Differences Persist in the Placenta Revealing Widespread Transient Imprinting. Plos Genet. 2016; 12: e1006427. [CrossRef] [Google scholar] [PubMed]
- Schroeder C, Sturm M, Dufke A, Mauholzmann U, Eggermann T, Poths S, et al. UPDtool: a tool for detection of iso- and heterodisomy in parent-child trios using SNP microarrays. Bioinformatics. 2013; 29: 1562-1564. [CrossRef] [Google scholar] [PubMed]

