

The Feasibility and Necessity of a Revolution in Geriatric Medicine
Giacinto Libertini ![]()
- Italian Society for Evolutionary Biology, 14100 Asti, Italy
- Department of Translational Medical Sciences, Federico II University, 80138 Naples, Italy
Received: February 20, 2017 | Accepted: April 12, 2017 | Published: April 17, 2017
OBM Geriatrics 2017, Volume 1, Issue 2, doi:10.21926/obm.geriat.1702002
Academic Editor: Michael Fossel
Recommended citation: Libertini G. The Feasibility and Necessity of a Revolution in Geriatric Medicine Giacinto Libertini. OBM Geriatrics 2017;1(2):002; doi:10.21926/obm.geriatr.1702002.
© 2017 by the author. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Currently, geriatric medicine consists mainly of palliative treatment of the disorders that characterize senile decay. This is perfectly compatible with the prevailing view that aging is the inevitable result of multiple degenerative processes that cannot be treated effectively as they are, in themselves, inevitable and irreversible.
This interpretation of aging clashes with a mass of data and arguments that, conversely, indicate that aging is a specific physiological function, favoured by supra-individual natural selection, and is genetically determined and modulated. According to this concept, it is possible to modify, or even reverse, aging by influencing its primary mechanisms. This is diametrically opposed to the current interventions used in geriatric medicine, which act only on the effects of such mechanisms.
The goal of complete control of the aging process may appear Utopian; however, it is quite rational and feasible if we consider the already proven reversibility of aging at the cellular level and in some in vivo models. The method of achieving this objective by no means is to counter the countless changes that characterize aging. On the contrary, the method is to control the telomere-subtelomere-telomerase system, which appears to be the general determining factor and regulator of aging. This system can apparently be controlled by modifying telomerase activity, or even, as suggested recently, in combination with techniques that alter telomere and subtelomere structure.
In the first instance, these interventions must predominantly address the control of some manifestations of aging, such as Alzheimer’s disease, Parkinson’s disease and age-related macular degeneration, which are particularly devastating in terms of patient suffering and the associated economic burden. Effective treatments for these diseases represent a pivotal challenge facing a revolution in geriatric medicine, which is now both feasible and essential. The implications of such a revolution will lead to developments that will extend well beyond the boundaries of geriatric medicine.
Keywords
Aging; Telomere; Subtelomere; Telomerase; Alzheimer’s Disease; Parkinson’s Disease; Age-Related Macular Degeneration
Introduction
It is impossible to discuss geriatric medicine, i.e., the prevention and care of the manifestations of senile decay, without precise scientific knowledge of the aging process. First, a clear definition of aging that does not contain, clearly or covertly, pre-conceived ideas about its primary causes is indispensable.
A neutral definition of aging is: “increasing mortality [i.e., fitness decline] with increasing chronological age in populations in the wild” [1], which may be well summed up as “actuarial senescence” [2], if the necessary specification “in the wild” is added.
Definitions of aging include: “a persistent decline in the age-specific fitness components of an organism due to internal physiological deterioration”[3], “progressive loss of function accompanied by decreasing fertility and increasing mortality with advancing age” [4], and “age-dependent decline in physiological function, demographically manifest as decreased survival and fecundity with increasing age” [5]. These definitions are also acceptable if “evident physiological degeneration” is not considered a synonym of “increasing mortality”, as an increased death rate may also occur with a slight reduction in fitness without any evident physiological degeneration. This question is not trivial. Indeed, if, under natural conditions, rare individuals with overt physiological alterations are sought, i.e., in a condition definable as a “state of senility” [6], aging will be a rarity in the wild (“aging is extremely difficult to observe in the natural habitats of most organisms” [3]). In this sense, aging would be irrelevant in natural selection [4].
In 1957, Williams wrote: “Comfort is severely critical of Weissmann’s theory [who proposed that senescence was favoured by natural selection], and offers in its place the theory that senescence is selectively irrelevant. He argues (e.g., 1956: 39) that senescence is outside the developmental program that concerns natural selection, since almost no wild organisms ever attain the senile stage. I believe that this theory is incorrect. Its fallacy lies in the confusion of the process of senescence with the state of senility ... No one would consider a man in his thirties senile, yet according to athletic records and life tables, senescence is rampant during this decade.” [6] This is a longstanding and important error. Indeed, a recent review has documented evidence of aging, defined as increasing mortality with chronological age in populations in the wild, in 175 species [7]. Moreover, the existence of species with evident aging under natural conditions, including humans [8], has been known for a long time [1,9,10]. Thus, aging cannot be considered irrelevant in natural selection.
Many theories have been proposed to explain the causes of aging [11,12,13]. These theories can be divided into two opposing categories of interpretations [14,15] and, in the sense proposed by Kuhn [16], are worthy of being defined as paradigms for their diversity and the importance of their implications.
The “non-programmed aging” paradigm maintains that: (i) aging is harmful to the individual and so is unlikely to be favoured by natural selection (“any hypothetical ‘accelerated ageing gene’ would be disadvantageous to the individual. It is, therefore, difficult to see how genes for accelerated ageing could be maintained in stable equilibrium, as individuals in whom the genes were inactivated by mutation would enjoy a selection advantage.” [4]); (ii) aging is the effect of many inevitable damaging factors that, for various reasons, natural selection may counter only partially [4]. Within this paradigm, the mechanisms of natural selection are disregarded or scarcely considered in older theories [11], while some more recent theories are based on the hypothesis that the damaging factors are insufficiently opposed by natural selection because: (i) in the wild, there are few “old” individuals and so natural selection against aging is weak (mutation accumulation hypothesis) [17,18]; (ii) there are genes with pleiotropic effects, i.e., advantageous in the young and deleterious in “older” individuals (antagonistic pleiotropy hypothesis) [3,6]; and (iii) there are limits determined by other physiological necessities (disposable soma hypothesis) [19,20]. For this paradigm, aging is a useful term used only to summarize many distinct phenomena and is not a unitary phenomenon. Therefore, as this is now the prevailing paradigm, in the current international classifications of diseases (ICD9 [21] and ICD10 [22]), there is no distinct code for aging and, so, the World Health Organization cannot provide statistics for aging as a cause of death [23].
In contrast, the “programmed aging” paradigm maintains that aging is a physiological phenomenon, i.e., something genetically determined and regulated that is favoured by supra-individual natural selection [15]. According to this hypothesis, aging is certainly harmful to the individual but, in principle, a gene that is detrimental to the individual may be favoured by natural selection at a supra-individual level. This is widely accepted for the many types of phenoptosis (“programmed death of an organism” [24]), which form a large category of well-known phenomena [9], although without a unifying definition until recently [25]. Within the programmed aging paradigm, there is an interpretation of aging proposed in terms of supra-individual selection. This interpretation explains aging as a beneficial phenomenon in populations spatially separated in demes, and in conditions of K-selection [26] because it accelerates the spread of any gene within the population. This was described by a kin selection mechanism [1] or using population models [27,28,29]. When the hypothesized conditions that favour aging are absent, the existence of non-aging species is expected. Indeed, the existence of species that show no age-related decline in fitness is well-known; these are defined tentatively as species with “negligible senescence” [9]. According to this paradigm, aging is a distinct phenomenon, the sole origin of which is concealed by its countless manifestations as well as the various manifestations of a disease (e.g., diabetes) might conceal its oneness.
These concepts are illustrated in Figure 1.
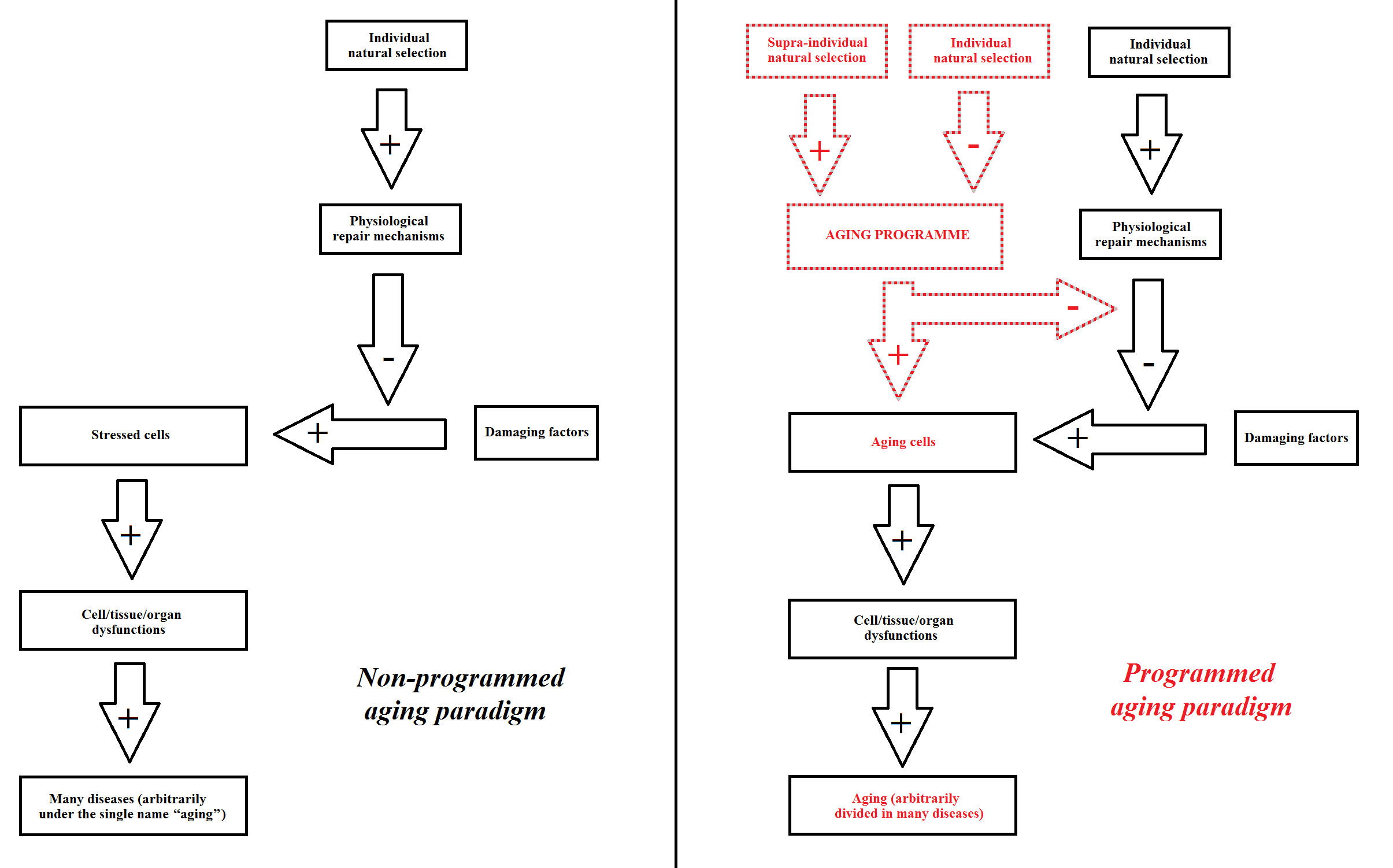
Figure 1 Interpretation of aging according to the non-programmed aging paradigm (on the left) and the programmed aging paradigm (on the right).
The main difference between the two paradigms can be summarized as follows:
- The “non-programmed aging” paradigm is based on aging being caused by several harmful factors. Therefore, this paradigm is not based on the existence of genetically determined and regulated mechanisms that cause aging and: “... had aging evolved in a non-adaptive way, it would have been impossible to provide justification for any well-defined program” [30]. Indeed, such mechanisms, if any exist, would prove that the paradigm is false. However, if this paradigm is true, any action against aging should attempt to minimize the effects and consequences of unavoidable damaging factors.
- In contrast, the “programmed aging” paradigm implies the existence of these mechanisms, and their absence would demonstrate that the paradigm is false. However, if this paradigm is true, any action against aging should be aimed at modulating or blocking the aforementioned mechanisms.
This difference is clearly essential to the determination of possible actions aimed at countering aging. Therefore, further elucidation of the mechanisms proposed by the programmed aging thesis is well within the objectives of this review. However, other issues that are not directly relevant to this goal will be omitted. In particular, we will not debate the evidence and arguments in support of the programmed aging thesis and/or against the opposite view, as this has been discussed elsewhere [15]. Neither will be discussed the phylogeny of aging [13], while the pathology of aging, which is outlined elsewhere [31,32,33], will be described summarily where necessary.
The immediate objection to this approach is that the choice to follow only the programmed aging paradigm is questionable and biased. However, the answer is simple and straightforward: in this review, the aforementioned paradigm is considered as a working hypothesis, while in many other reports the opposite view is accepted, implicitly or explicitly, as a working hypothesis. In accordance with good scientific practice and beyond the theoretical arguments and the evidence accumulated so far, the results of these evaluations will reveal the most useful working hypothesis.
Aging Machinery: The Telomere Theory
Although other descriptions have been proposed for the mechanism of aging (e.g., [34,35,36,37,38]), here, we consider and describe the essentials of only the most homogeneous theory that is supported by evidence; for brevity this will be defined as “telomere theory”. References to other reviews [32,33,39,40] will be made to provide a more detailed description and a more extensive list of the reports on which it is based.
The Telomere-Subtelomere-Telomerase System
At each end of the chromosomal DNA molecule, there is a region with a repetitive sequence called the telomere. This sequence, which is TTAGGG in humans and other mammals [41], is highly conserved and is present in many phylogenetically distant species [42].
Between the telomere and the main part of the DNA molecule, there is another region, the subtelomere (functionally defined below), with an “unusual structure: patchworks of blocks that are duplicated” [43]. “A common feature associated with subtelomeric regions in different eukaryotes is the presence of long arrays of tandemly repeated satellite sequences.” [44].
The telomere is covered by a heterochromatin hood [39] (Figure 2).
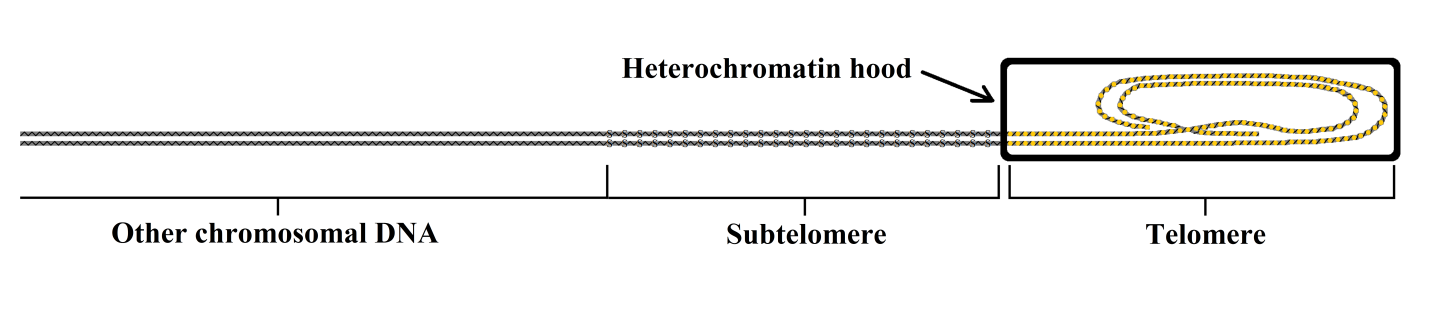
Figure 2 Schematic representation of the structures of the telomere, its heterochromatin hood, and the subtelomere.
With each replication, a small part of the telomere is left unduplicated by the DNA polymerase [45,46]. As excessive shortening of the telomere would jeopardize cell vitality, the existence of an enzyme with the capacity to restore the unduplicated part was predicted [47]. This enzyme, telomerase, was identified in 1985 [48], thus explaining the capacity of stem and germ line cells to duplicate numerous or even unlimited times. Later, telomerase activity was shown to be repressed by specific regulatory proteins [49]. Moreover, there is an age-related progressive shortening of telomeres in many cell types [50] and an association between telomere length of an individual and its life expectancy has been identified in animal species studied under natural conditions [51,52,53]. This explained the limits of cell duplication capacities demonstrated both in vitro [54,55,56,57,58] and in vivo [59], as well as the inverse relationship between duplication capacities and age among different individuals [60], and the direct relationship between duplication capacity and longevity among different species [61].
However, studies of cultured cells that had undergone an equal number of previous replications showed that cell duplication ceased at a random time, but with an increasing probability related to telomere shortening. This caused a progressive decrease in the average duplication capacity of the culture as a whole, but without a concomitant inability of individual cells to duplicate after a certain number of replications [62,63]. This phenomenon was later explained [64] by the observation that the telomere is “capped” by the heterochromatin hood but, at some times, the hood is temporarily detached and the telomere is “uncapped” for a period related to telomere shortening. In this phase, the cell is vulnerable to the triggering of “cell senescence”, which is a “fundamental cellular program” [65] that blocks the cell replication capacity (“replicative senescence”). Moreover, even when the telomere is minimally shortened, there is a short “uncapped” phase and so, a small probability of triggering cell senescence.
Another phenomenon occurs as a consequence of telomere shortening. When the telomere shortens, the heterochromatin hood slides over the subtelomere and this determines the transcriptional silencing of the subtelomere covered region [39]. This silencing effect, known as the “telomere position effect” [66] and defined also as “gradual senescence” [13], determines an altered functioning of genes “over long distances” in the chromosome [67]. Moreover, there is an effect on many cell functions, including cellular secretions (e.g., collagen, elastin, etc.), leading to alterations in the intercellular matrix, inflammation and damage to other cells [39]. Gradual senescence allows a functional definition of the subtelomere as follows: one end is where the telomere begins (junction subtelomere-telomere), while the other end is the farthest subtelomere section repressed by the greatest sliding of the telomere hood [40].
These phenomena indicate that the subtelomere has important regulatory functions on: (i) many cell functions; and (ii) the “capped”/“uncapped” alternation, i.e., on the probability of triggering the cell senescence program. This is compatible with the presence of repeated sequences in the subtelomere, and the hypothesis that these sequences perform the aforementioned regulatory functions, which are progressively silenced by the sliding of the telomere hood (Figure 3).
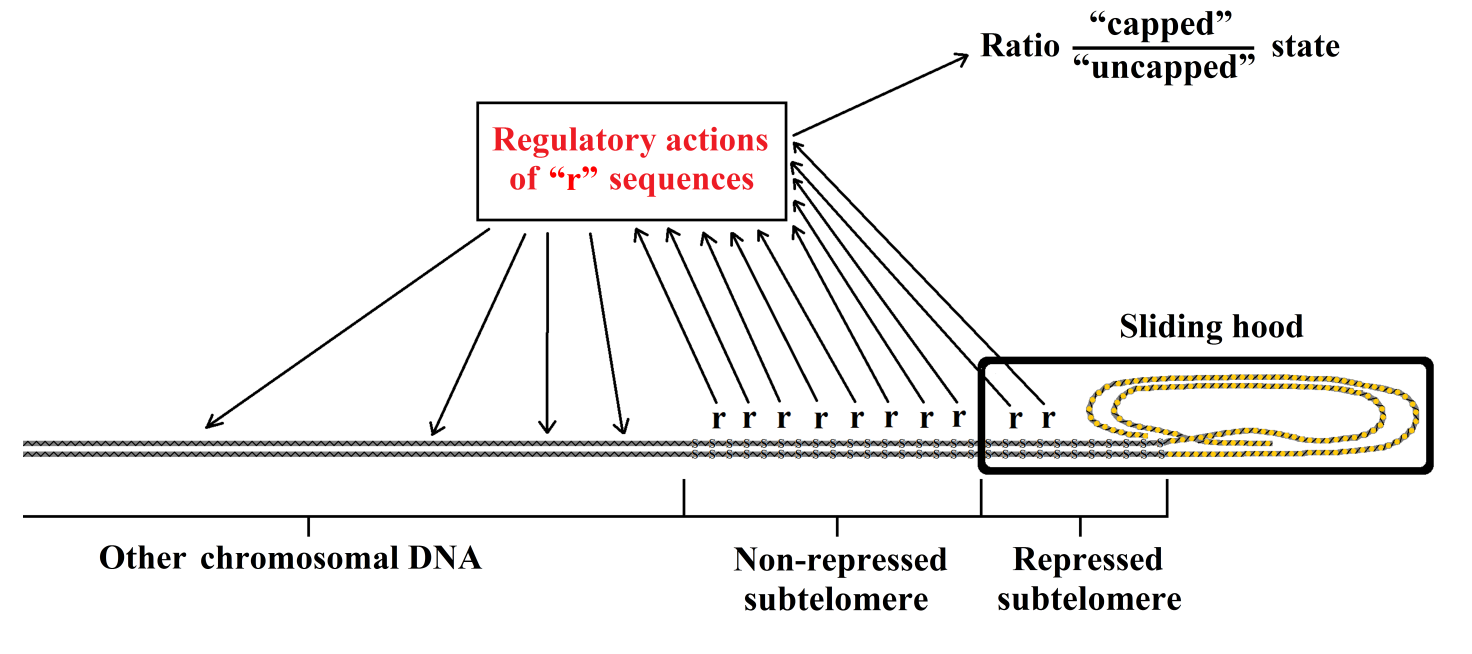
Figure 3 Schematic representation of telomere sequences (“r”) with regulatory functions and of their repression by sliding of the telomere hood.
Absence of Correlation between Longevity and Telomere Length in Germ Line Cells
A simple prediction is that, in different species, longevity should be related to telomere length in germ line cells (“initial telomere length”) and/or to the intensity of telomerase activity. However, the following evidence has clearly disproved this prediction:
(i) mice and hamsters have long telomeres but age precociously [68];
(ii) in different rodent species, longevity and telomerase activity are not related [69];
(iii) two Mus musculus strains with different telomere lengths showed the same lifespans and patterns of the timing of cell senescence [39];
(iv) donor animals and cloned animals derived from the somatic cells of the donors had different telomere lengths (shorter in the cloned animals), but showed the same senescence timing [39];
(iv) under protected laboratory conditions, mice with genetically inactivated telomerase showed impaired fertility and viability only after four to six generations, i.e., when there was a considerable telomere shortening [70,71]. However, as alterations were found in early generations in organs with high cell turnover [71,72], it is likely that, in the wild, fitness would be compromised even in the first generation;
(v) mice had limited longevity, but showed baseline telomerase activity in most somatic cells [73].
A detailed account of a possible explanation for these phenomena, which are apparently inconsistent with the telomere theory, has been presented elsewhere [40], and for brevity will not be repeated here. However, in summary:
(i) the germ cell – in a period definable as the “reset phase” – forms the heterochromatin hood with a size proportional to the length of the telomere. Regarding longevity, the absolute “telomere length in this phase is irrelevant” [39], except when telomere length is below a critical value [39];
(ii) the heterochromatin hood necessarily has a fixed length in all cells of the organism that are derived by duplication from the germ cell;
(iii) with each duplication, the telomere shortens and the adjacent subtelomeric region is gradually repressed by sliding of the heterochromatin hood;
(iv) the critical factor is not the absolute initial telomere length, but the “relative” telomere shortening and therefore, the fraction of the subtelomere that is repressed [39];
(iv) greater telomerase activity would slow down telomere shortening, and thus, result in slower aging, although this could be balanced, or even tipped in favour of more rapid aging, by a shorter subtelomere, leading to repression of a greater fraction of the subtelomere. Under such conditions, it is possible that species exist with longer telomeres and greater telomerase activity, yet with shorter longevity, as is the case in mice.
It is important to consider the unicellular species yeast (Saccharomyces cerevisiae), which exhibits some characteristics that may be considered to represent aging. Each yeast cell divides into two cells, which are defined as “mother” and “daughter” cells. While the cells of the daughter lineage are identical to the parent cell, replication of the cells of the mother lineage is limited (approximately 25–35 duplications [74]). In relation to the number of duplications, there are increasing metabolic alterations [75,76,77,78,79] and growing vulnerability to death by apoptosis and replicative senescence [75,76,78,79]. These phenomena easily explain the mortality differences among individual yeast cells under particular conditions of stress. However, among the cells of the mother lineage, the mortality rate increases with an exponential dynamic related to the number of duplications [80]. This characteristic is similar to the age-related increase in mortality, i.e., aging, shown by many multicellular species [7,10]. Thus, these phenomena may be considered within the concept of aging as previously defined [1].
In both mother and daughter lineage cells, telomerase is constitutively active and there is no telomere shortening with each duplication [81,82,83]. However, in the cells of the mother lineage, accumulation of particular molecules (extrachromosomal ribosomal DNA circles [ERCs]) occurs over the subtelomere [84]) in a manner that is proportional to the number of duplications, and which alter cell viability progressively [77]. In yeast tlc1Δ mutants, telomerase is inactive and telomeres shorten with each replication both in mother and daughter lineage cells. Although cells of the daughter lineage does not undergo the ERC accumulation that occurs in the mother cells, they do show the same alterations and the same overall expression of genes (i.e., the transcriptome) as mother lineage cells with the same number of duplications [77]. However, an interesting analogy must be highlighted: (i) the daughter cells of the normal strains are similar to those of the germ line of multicellular eukaryotes, i.e., active telomerase, no telomere shortening, no sliding of the heterochromatin hood over the subtelomere, no repression of the subtelomere, no alteration of cell functions; while (ii) the daughter cells of tlc1Δ mutants are similar to non-germ line cells of multicellular eukaryotes after some duplication, i.e., reduced or absent telomerase activity, telomere shortening, sliding of the heterochromatin hood over the subtelomere, repression of part of the subtelomere, and altered cell functions (Figure 4).
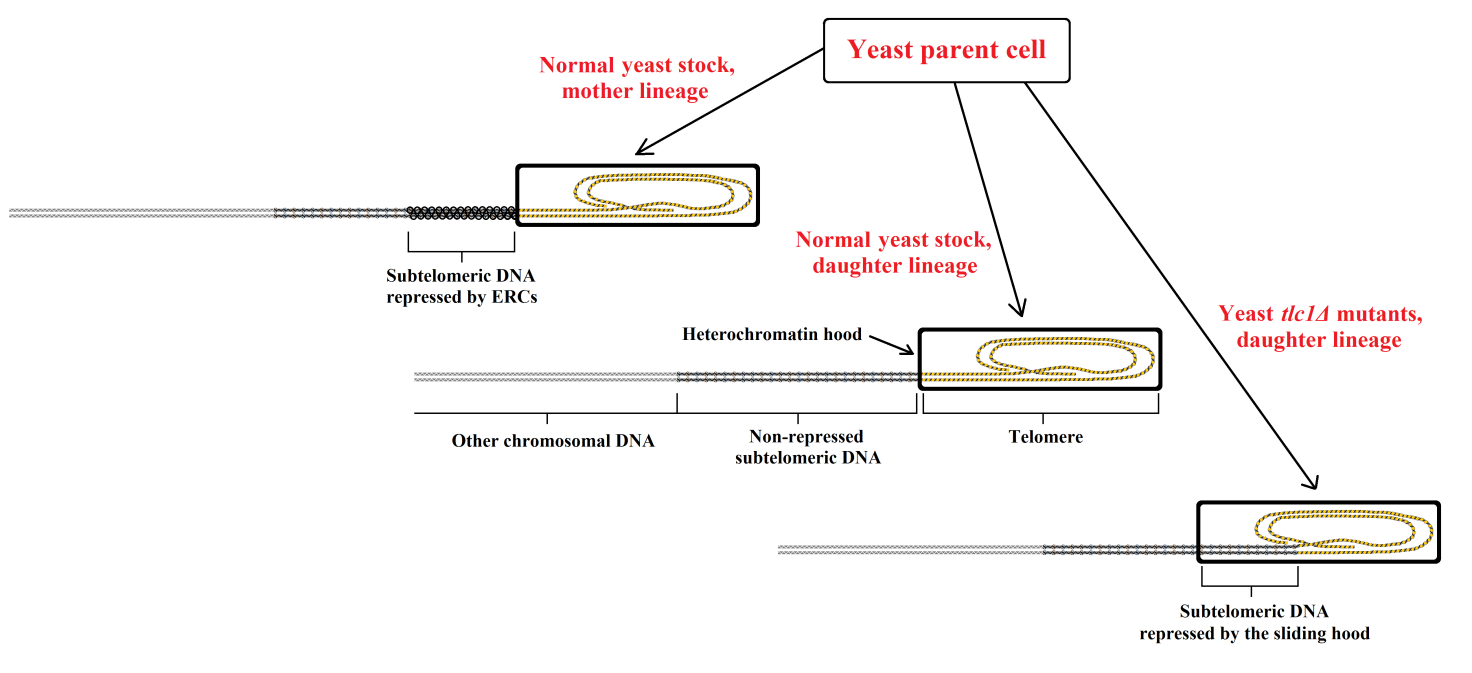
Figure 4 In normal yeast stocks, the condition of daughter cells is similar to that of the germ line cells of multicellular eukaryotes, while in yeast tlc1Δ mutants, it is similar to that of the non-germ line cells of multicellular eukaryotes after some duplication.
Cell Turnover
In the absence of accidental events that cause cell necrosis (trauma, ischaemia, infection, etc.), in general, cells die by programmed cell death (PCD). Examples of forms of PCD include: (i) the detachment of cells from the intestine and other internal walls of body cavities; (ii) the keratinization and detachment of the epidermis and hair cells; (iii) osteocytes eliminated by osteoclasts; (iv) erythroblasts transformed into erythrocytes and later removed by macrophages; (v) apoptosis, an orderly process of self-destruction where cell debris are removed and reused without damage to other cells.
Apoptosis, a phenomenon described for the first time in normal hepatocytes in 1972 [85], is phylogenetically ancient (e.g., a form of apoptosis is observable in S. cerevisiae [80,86]). Moreover, disregarding other functions, in vertebrates apoptosis is essential for cell turnover in normal adult organs [87,88,89] and is observed for many tissues/organs (e.g., gliocytes [62], liver [90], thyroid [91], pancreatic β-cells [92], adipocytes [93], skeletal muscle [94,95], bone [96], cartilage [97], kidneys [98], biliary epithelial cells [99], prostate [100], and lung type II alveolar epithelial cells [101]).
Cell death, caused by apoptosis and other types of PCD, is a continuous process that is balanced by equivalent production of new cells by duplication of stem cells that are specific for each cell type. The pace of this continuous cell turnover varies greatly depending on the cell type and tissue [102]. For example, osteocytes are renewed approximately every 10 years [103] and heart myocytes have a turnover of approximately 4.5 years [104], while cells of the intestinal epithelium are renewed every 3–6 days [103]. The total number of cells that die and are replaced is impressive. It is estimated that, in one year, the turnover is approximately equal to the mass of the whole organism and that 50–70 billion cells die and are substituted every day [105].
The Atrophic Syndrome
The progressive telomere shortening with each cell duplication determines:
(i)an increase in gradual senescence; and
(i)an increasing probability of the activation of cell senescence (replicative senescence and gradual senescence in the highest degree).
The effects of telomere shortening on cell functions and cell turnover include:
(i) an increase in the number of cells with functions altered in varying degrees by the gradual senescence;
(ii) an increase in the number of cells in the senescent state, i.e., without replicative capacity and with gradual senescence in the highest degree;
(iii) a reduction in the number of stem cells that can replicate, and so a reduction in cell turnover;
(iv) a reduced number of cells (atrophy);
(v) a substitution of lost specific cells with non-specific cells;
(vi) a compensatory hypertrophy of the remaining specific cells;
(vii) alterations in the intercellular fluids and substances;
(viii) alterations in the cells depending on the functionality of the lost cells or of the cells in gradual senescence;
(ix) greater vulnerability to cancer because of dysfunctional telomere-induced instability [106];
(x) a general decline in the functions of cells/tissues/organs, i.e., progressive fitness decline, i.e., aging.
This condition has been defined as “atrophic syndrome” [31,32,33] and describes the progressive anatomical and functional alterations that characterize aging. The description of the “atrophic syndrome”, which contrasts with the interpretation of the non-programmed aging paradigm, is analogous to descriptions expressed by others: “The accumulation of dysfunctional cells, together with a limited regenerative capacity of tissues, is thought to determine the age-related decline of body organs ... Dysfunctional cells, usually characterized by the presence of short telomeres ...” [107].
A schematic representation of atrophic syndrome for cell types that are subject to turnover (“direct aging”) is shown in Figure 5.
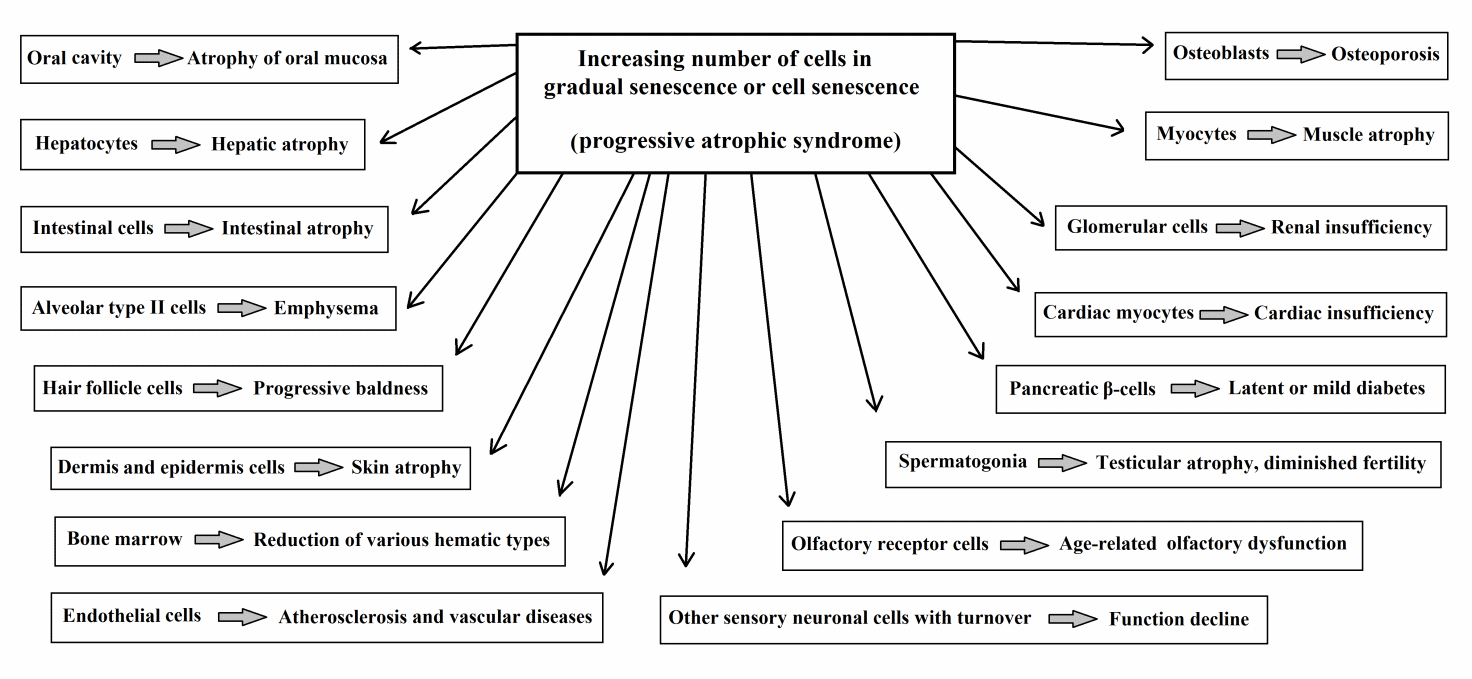
Figure 5 Telomere theory: atrophic syndrome for cell types that are subject to turnover (“direct aging”).
Cell Turnover and Perennial Cells
The general rule of cell turnover has some important exceptions.
With some exceptions [108,109], central nervous system neurons do not undergo cell turnover. This is due to the connection of these cells to other neurons in a variable manner via numerous synapses. It is estimated that there are 1011 neurons connected by 1014 synapses in a human brain [110], which means that each neuron is connected with an average of 1,000 synapses. The replacement of these neurons with new elements would require an improbable level of fidelity in the restoration of the connections of the replaced neurons. Conversely, where this problem does not exist, neurons do undergo turnover. For example, olfactory receptor cells, which are specialized neurosensory neurons, receive a molecular signal from the external environment via a single dendrite, which is then transmitted to other neurons by a single axon [111]; turnover in this cell type is well-known [112].
However, neurons that do not undergo turnover depend on other cells that are subject to turnover for their vitality. For example, retinal photoreceptor cells need the macrophagic activity of retinal pigmented epithelium cells, which are highly differentiated gliocytes that undergo turnover [113,114]. The decline in these satellite cells determines the accumulation of damaging substances, such as A2E (a breakdown product derived from vitamin A [115]), photoreceptor death, and therefore progressive manifestations of age-related macular degeneration (AMD) [113,116].
The pathogenesis of Alzheimer’s disease (AD) has also been explained by the decline of satellite glial cells [31,32,39,117,118]. Similar mechanisms have been proposed for Parkinson’s disease (PD) and senile presbycusis [118].
The crystalline lens, which is a particular case analogous to that of perennial cells, has been discussed elsewhere [118]. The lens lacks nucleated cells in its core and aspects of its functionality, including transparency, depend on lens epithelial cells that undergo turnover [58]. The age-related reduction in the duplication capacity of lens epithelial cells in healthy human subjects is well-known [58], and “It is apparent in Werner syndrome that the cataracts result from alterations in the lens epithelial cells” [119].
A schematic representation of atrophic syndrome in cell types and organ parts without turnover (“indirect aging”) is shown in Table 1 and Figure 6.
Table 1 Atrophic syndrome in cell types and organ parts without turnover (“indirect aging”).
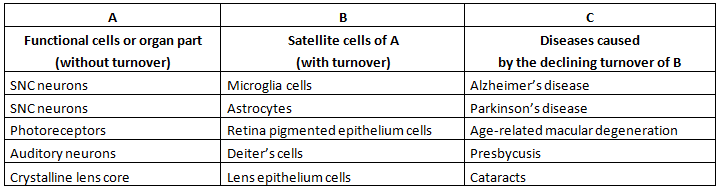
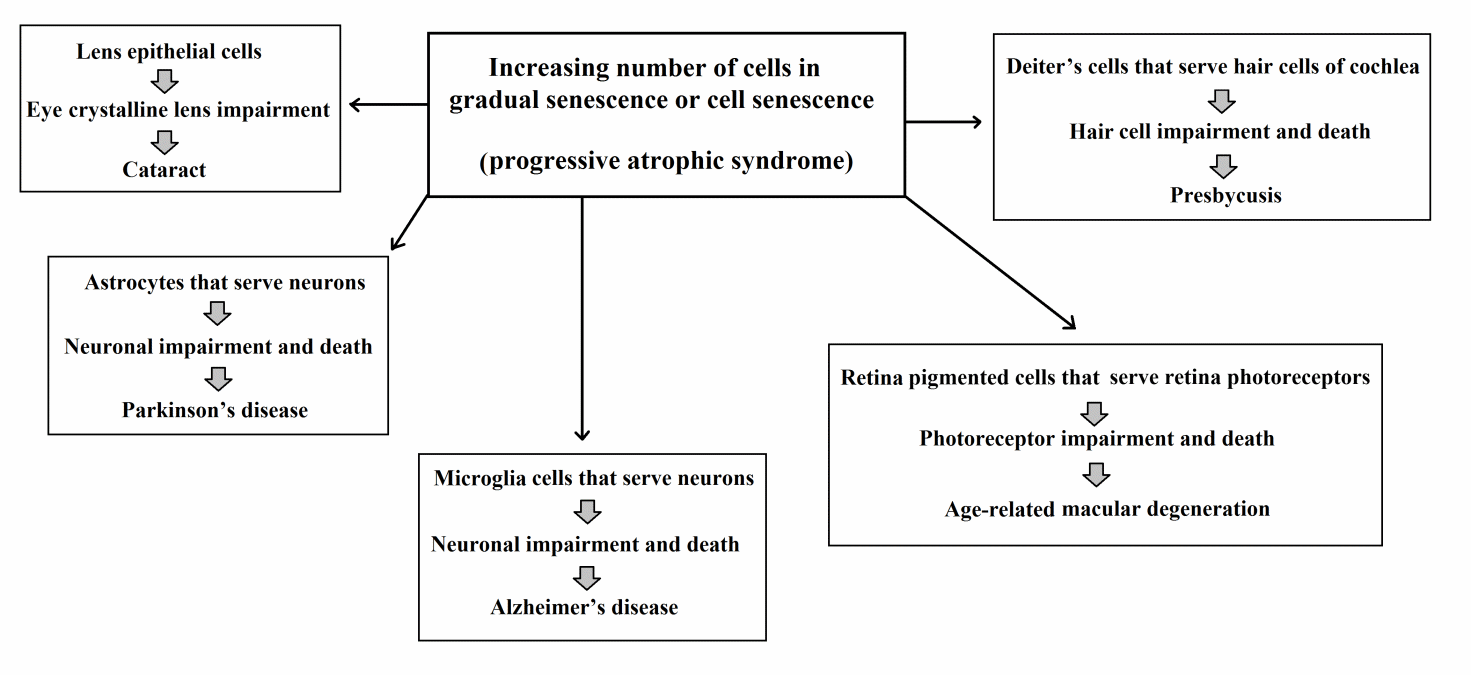
Figure 6 Telomere theory: atrophic syndrome in cell types and organ parts without turnover (“indirect aging”).
A detailed description of atrophic syndrome in each tissue or organ is beyond the scope of this review. A partial and brief description has been reported elsewhere [31,32,33] and only a few concise descriptions will be presented here as examples.
- Liver. Liver volume shows an age-related decline, estimated to be approximately 37% between the ages of 24 and 91 [120], both in absolute values and in proportion to body weight [121]. In elderly individuals, the size of the remaining hepatocytes increases, while hepatocyte size declines in liver atrophy caused by starvation [122,123].
- Heart. In contrast with long-held and deep-rooted convictions, the heart is a self-renewing organ. In a normal human heart, approximately 3 million myocytes die by apoptosis each day and are replaced by cardiac stem cells. In fact: “the entire cell population of the heart is replaced approximately every 4.5 years … The human heart replaces completely its myocyte population about 18 time during the course of life, independently from cardiac diseases.” [104]. Cardiac stem cells allow myocyte turnover and exhibit age-related telomeric shortening and cell senescence [124,125,126]. The heart shows an age-related decline in the number of myocytes and an increase in cell volume per nucleus in the remaining myocytes [127]. The loss of myocytes causes a decline in cardiac dynamic capacities and this causes enlargement of the heart, i.e., a morphological hypertrophy, which conceals the underlying atrophy in the number of contractile cells [128]. “With aging, there is also a progressive reduction in the number of pacemaker cells in the sinus node, with 10 percent of the number of cells present at age 20 remaining at age 75. ... Age-associated left ventricular hypertrophy is caused by an increase in the volume, but not in the number of cardiac myocytes. Fibroblasts undergo hyperplasia, and collagen is deposited in the myocardial interstitium.” [128].
- Skin. In older individuals, the dermal-epidermal junction, where the stem cells for the epidermis are located, is flattened. Furthermore, all the components of the derma (dermal fibroblasts, melanocytes, mast cells, Langerhans cells, eccrine glands, capillaries, Pacinian and Meissner’s corpuscles, hair, etc.) decrease in number; and nails are reported to grow more slowly [129].
- Endothelium. Endothelial cells show cell turnover and their replacement is ensured by stem cells known as endothelial progenitor cells (EPCs) and derived from primary stem cells in the bone-marrow. The number of EPCs decreases with age and due to the effects of “cardiovascular risk factors” (diabetes, cigarette smoking, hypercholesterolemia, hypertension, etc.), while the number is increased by statins and other drugs [130]. A reduced number of EPCs decreases endothelial cell turnover and jeopardizes endothelial function, which alters blood circulation and causes diseases such as cardiac infarction or cerebral ischaemia. The decline in EPC number is a reliable cardiovascular risk index, equivalent to the Framingham risk score [130,131]. In older individuals, excluding the effects of cardiovascular risk factors, the incidence of diseases caused by altered endothelial function increases exponentially [132].
Reversibility of the Manifestations of Aging
If aging is caused by the accumulation of inevitable damage of various kinds, as is maintained in general by the non-programmed aging paradigm, then, in principle it should be, at least in part, irreversible. In contrast, if aging is determined by mechanisms that are genetically determined and modulated, as is maintained by the programmed aging paradigm, then, in principle it could be regulated or even reversed. There are several examples in clear support of the second hypothesis and that undermine the opposite hypothesis. These include: (i) in vitro, telomerase activation cancels the cellular biochemical alterations associated with gradual senescence and cell senescence [133,134,135,136,137]; (ii) fibroblasts obtained from the skin of young human donors were found – by the methods used – to be indistinguishable from those obtained from older donors with reactivated telomerase [138]; (iii) in aged mice in which telomerase was genetically blocked, telomerase re-activation reversed the manifestations of aging, even those of the nervous system [139]; and (iv) telomerase re-activation, in 1- and 2-year-old normal mice, delayed the manifestations of aging and increased the lifespan [140].
The Telomere-Subtelomere-Telomerase System and Cancer
The non-programmed aging paradigm precludes the existence of genetically determined and modulated mechanisms, which cause progressive fitness decline (i.e., the telomere-subtelomere-telomerase system), since natural selection could not allow their evolution and, if existing, would eliminate them. In any case, these mechanisms are incompatible with this paradigm [141]. Contrary to these assertions, the evidence for the aforementioned mechanisms appears indisputable, although their existence is still implicitly disregarded [142], or overtly denied, both in principle and ignoring precise previous descriptions [31,32]. For instance: “Aging is not and cannot be, programmed ... Programmed theories neither specify nor predict mechanisms of death” [143]; “... biological aging can be defined as the random, systemic accumulation of dysfunctional molecules that exceeds repair or replacement capacity ... aging is not a programmed process, it is not governed directly by genes. On the contrary, aging is a stochastic process” [144].
However, authoritative researchers (who have contributed to the detection of these mechanisms through remarkable studies) believe that, to justify their existence, there is the absolute necessity of an important function for these mechanisms that must be distinct from that of lowering fitness and must overcome, in terms of natural selection, the clear damage caused by their action. The only proposed explanation that is compatible with the non-programmed aging paradigm suggests that these mechanisms are a general defence against cancer, since replicative senescence would diminish or even block cancer proliferation [145,146]. A deadly evolutionary trade-off between the need for defence against cancer and the manifestations of aging was therefore hypothesized [147], which was compatible with two old and popular theories (antagonistic pleiotropy theory [3,6] and disposable soma theory [19,20]).
However, this proposal is untenable as already explained elsewhere [31,40,148,149]. However, in short, the reasons are as follows:
(i) In yeast, gradual senescence, replicative senescence and vulnerability to apoptosis, caused by ERC accumulation, which has the same effects of telomere shortening in multicellular eukaryotes, are well-documented [74,80,150]. However, this does not protect against cancer, which is impossible in unicellular species, such as yeast.
(ii) Gradual senescence weakens cells and the organism as a whole. Moreover, when cell senescence is activated, there is also a greater resistance to apoptosis [151]. This is in contrast to the situation in yeast, in which a greater vulnerability to apoptosis increases the death rate. However, in multicellular eukaryotes, a greater resistance of senescent cells to apoptosis weakens the organism and so, increases the death rate. Therefore, the effect is the same in terms of the elimination of aged individuals. These phenomena are implausible as defences against cancer for two reasons: (a) in mice, selective elimination of senescent cells leads to an increased lifespan, fewer age-dependent changes, and a delay in the progression of cancerous diseases [152]; and (b) increasing numbers of cells in gradual senescence and replicative senescence progressively reduces the efficiency of the immune system [39], which should increase vulnerability to cancer and its incidence [153].
(iii) Animal species that show no age-related decline in fitness (“animals with negligible senescence” [9]) cannot have any age-related increase in vulnerability to neoplastic disease, as confirmed by their constant death rates at any age. However, in the wild, old individuals of these species have the same telomerase activity as young individuals [154,155].
(iv) Shortened telomeres cause DNA instability that increases the probability of cancer [106,156,157].
(v) A study on a human population in the wild showed that: (a) there was an age-related increase in mortality that began at age 30 (equivalent to that observed among the general population [6]); (b) approximately 30% of the population survived to 60 years of age and 20% to 70 years; and (c) no cases of cancer were reported and sporadic deaths due to cancer were only possible in individuals older than 70 years [8]. It appears illogical that the mechanisms underlying an age-related increase in mortality beginning at age 30 act as a defence against a rare disease found in much older individuals [148].
(vi) As telomerase activation, a common feature in cancer, occurs as a consequence of cancer and does not precede disease onset, it cannot be considered to be a causative event and should be considered only as a characteristic of cancerous cells that exacerbates the disease [39].
Therefore: “The hypothesis that telomerase is restricted to achieve a net increase in lifespan via cancer prevention is certainly false. Were it not for the unthinkability of the alternative – programmed death – the theory would be dead in the water.” [149]
However, the rejection of the hypothesis that the telomere-subtelomere-telomerase system is a defence against cancer does not exclude the possibility that aberrant telomerase activation could be carcinogenic under particular circumstances [158,159].
Pathology of Aging
If aging is a function, alterations in this function, viz. specific diseases, are foreseeable. Indeed, there are two main categories of diseases that affect aging:
(1) Diseases Caused by Genetic Alterations
These are rare but of great interest in investigations of aging. Two syndromes, dyskeratosis congenita (DC) [160] and Werner’s syndrome (WS) [119], are particularly interesting in this regard.
The autosomal dominant form of DC is caused by a defect in the gene that encodes the RNA part of telomerase [161], while the X-linked form of DC shows low levels of telomerase and shorter than normal telomeres [162]. “Problems tend to occur in tissues in which cells multiply rapidly – skin, nails, hair, gut and bone-marrow – with death usually occurring as a result of bone-marrow failure.” [163]
WS is caused by dysfunction in a RecQ family helicase that determines a dysfunction of somatic cells in the cycling state [164]. In WS, cells show high somatic mutation rates [165] and reduced replication capacity [60]. This abnormality in DNA metabolism causes an atrophic syndrome in non-high turnover cells and tissue [119].
The differences between the two syndromes have been carefully outlined [163] and were later discussed in the context of the programmed aging paradigm [32]. In short, DC and WS can be considered as two model cases of segmental progeria (i.e., the altered functionality of only a fraction of cell phenotypes) [39], and it is likely that a non-segmental (i.e., total) progeria is incompatible with life.
(2) Diseases Caused by Unhealthy Lifestyles
Unhealthy lifestyles (e.g., alcohol abuse, cigarette smoking and eating habits that increase the risk of hypertension, diabetes mellitus, and obesity) are defined overall as “risk factors”. These appear to increase the requirement for cell replication and so, accelerate aging. Conversely, healthy lifestyles and the use of drugs with organ protection qualities (“protective drugs”) are defined overall as “protective factors”. These appear to reduce the requirement for cell replication and to counter the accelerating effects of the “risk factors” on the aging process [31]. As a specific example, we have previously highlighted the existence of an inverse relationship between the number of EPCs and age and/or “cardiovascular risk factors” (i.e., factors defined previously as risk factors), while EPC number is increased by statins and other protective drugs [130]. These relationships are observable for many other manifestations of aging. In Table 2, for brevity, these relationships are considered in comparison with endothelial dysfunction only for AMD, AD, and PD.
Table 2 Relationships between some aging problems and some “risk factors” or “protective drugs”.
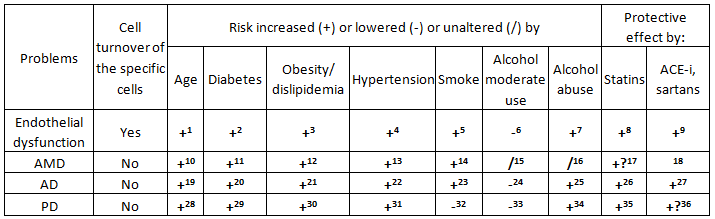
Notes: (1-5) [130,166]; (6) [167-169]; (7) [168,169]; (8) [170,171]; (9) [171]; (10) [172]; (11-14) [173,174];
(15) [175]; (16) [176]; (17) [177]; (18) No specific study; (19) [178]; (20) [179-183]; (21) [179,184];
(22) [179,185,186]; (23) [179,187]; (24) [188,189]; (25) [179,188]; (26) [184,190,191]; (27) [190-192];
(28) [193,194]; (29) [183,195-197]; (30) [197-199]; (31) [197]; (32) [200]; (33) [201]; (34) [202]; (35) [203-206];
(36) [207,208].
Differences between Non-Programmed and Programmed Aging Paradigms in the Definition of Aging Problems
According to the “non-programmed aging” paradigm, an elderly individual suffers, to varying degrees, from many different diseases (these are non-exhaustively summarized in Figures 5 and 6). Such diseases are caused by various inevitable harmful factors, although some of these factors may coincide for different diseases. These illnesses are not unique entities and therefore, the term “aging”, which is commonly used to define them together, must be considered as a lexical convenience that does not mean a scientific acceptance of their unit. The manifestations of each of these diseases may be anticipated and exacerbated, to varying degrees, by further damaging factors (“risk factors”). However, the limitation of these risk factors and specific treatments (“protective drugs”) may prevent, restrict or neutralize such additional damage. If we exclude any aggravating factor, these distinct diseases may be cured and counteracted only partially and with illusory and temporary effects. This is because these diseases are caused by factors that are intrinsic to organismal physiology, and inevitable and inexorable in their actions. Any treatment can only be applied to limit the damage suffered by cells and tissues, or, at a later stage, to limit or compensate for the damage that has occurred. This concept is summarized in Figure 7.
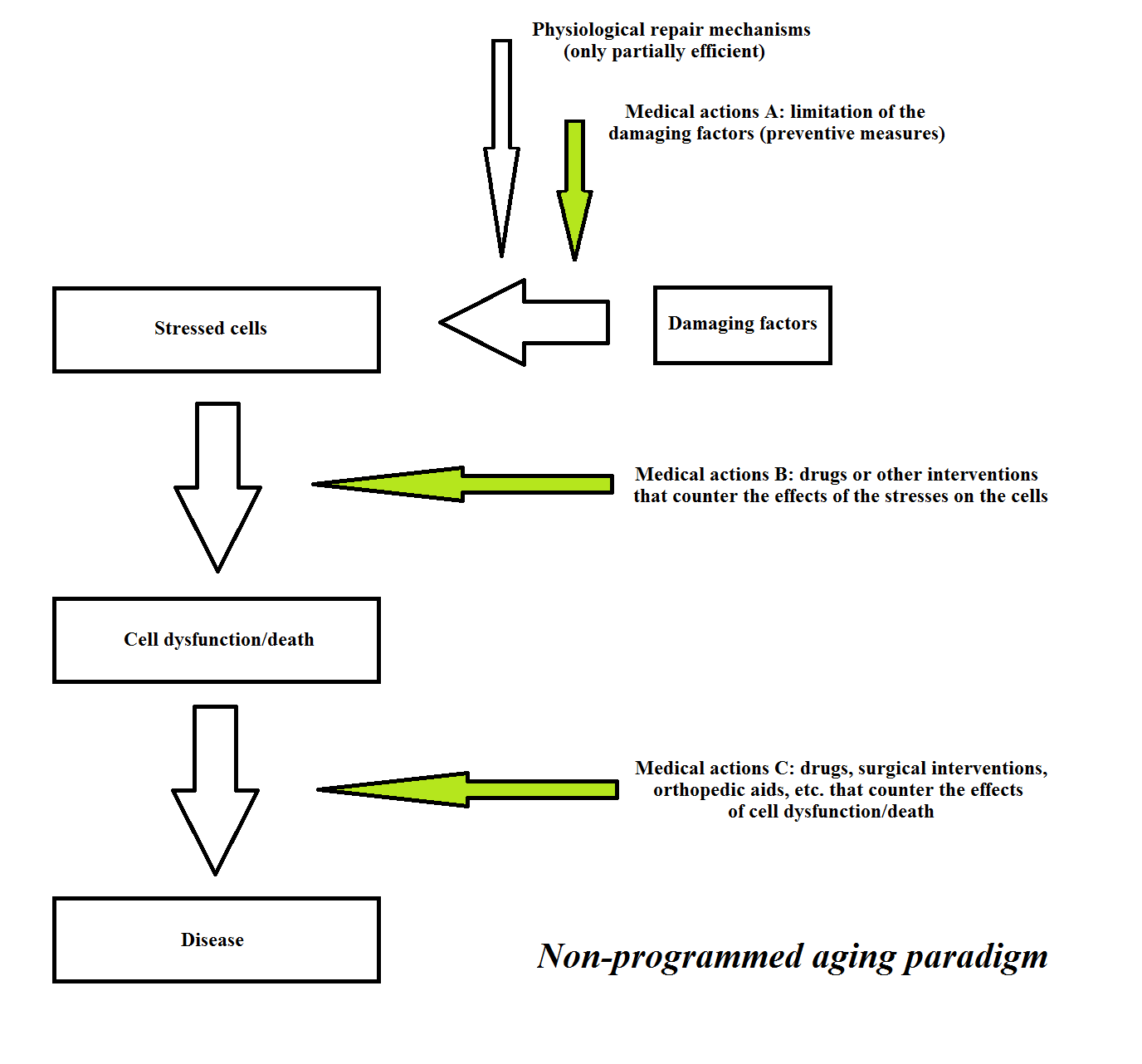
Figures 7 Traditional interpretation of the diseases that afflict the elderly. The concepts that underlie the present scheme are obtainable in any traditional textbook of geriatric medicine (e.g., [209]).
As an example, with reference to a single disease, Figure 8 illustrates the current approaches to the interpretation, prevention and care of AD.
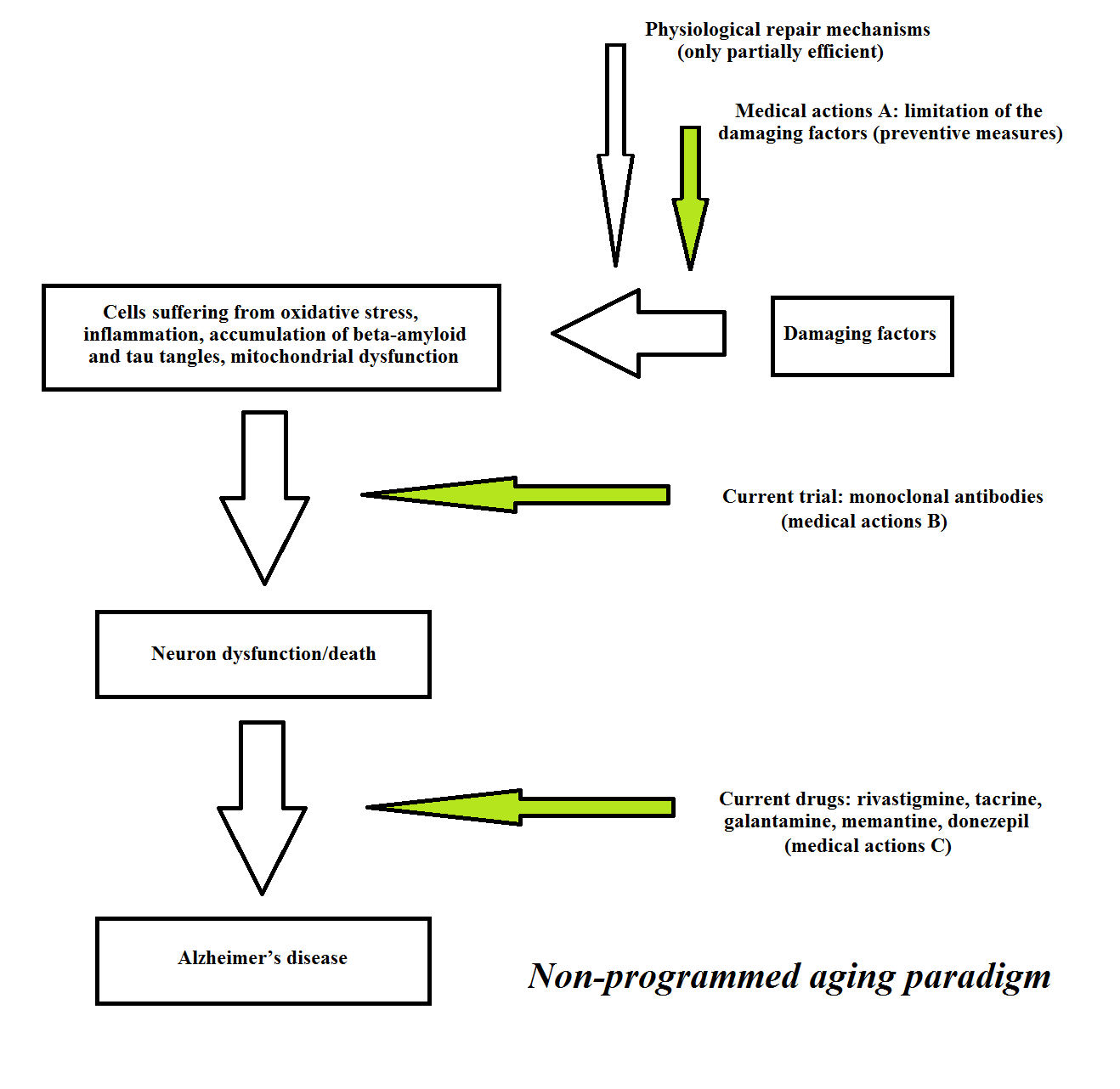
Figure 8 Current approaches to the interpretation, prevention and care of Alzheimer’s disease.
Regarding “medical actions B” for AD (see Figure 8), pharmaceutical companies have failed to identify an agent that will cure or halt the clinical manifestations of AD by the elimination of
β-amyloid and tau protein. In particular, studies of drugs or vaccines targeting the formation and accumulation of β-amyloid have yielded disappointing results [210]. One study showed that a vaccine could eliminate β-amyloid plaques and/or avoid their formation, without any positive clinical effects: “Seven of the eight immunised patients who underwent post-mortem assessment, including those with virtually complete plaque removal, had severe end stage dementia before death ... Although immunisation with Abeta(42) resulted in clearance of amyloid plaques in patients with Alzheimer’s disease, this clearance did not prevent progressive neurodegeneration” [211]. Another study showed that aducanumab, a fully human monoclonal antibody, was effective in eliminating β-amyloid plaques, although the clinical results were ambiguous [212].
Regarding “medical actions C”, (i) the neurological symptoms of AD are treated by acetylcholinesterase inhibitors (e.g., donezepil, galantamine, rivastigmine, and tacrine), N-Methyl-D-aspartate receptor antagonists (e.g., memantine), and other drugs [213,214]. However, “Current therapies for Alzheimer’s disease do not modify the course of disease and are not universally beneficial.” [213]; and (ii) Treatment of the cognitive alterations associated with AD is disappointing and, moreover, there is an increased long-term risk of mortality in relation to the use of antipsychotic drugs [215].
In short, “The only drugs available for Alzheimer’s patients aim to treat symptoms ... They are marginally effective at best.” [210].
In any case, according to the non-programmed aging paradigm, both the countless diseases, which in combination, constitute aging and the precocious and/or aggravated forms of the same diseases caused by unhealthy lifestyles or by genetic alterations, are always considered as individual diseases; therefore, their treatment is always considered to be a cure.
According to the programmed aging paradigm, the interpretation of the same problems is quite different. Aging is interpreted as a unique physiological phenomenon that manifests itself in various ways, depending on: (i) the tissue or organ concerned; (ii) previous life events; and, (iii) partially, random factors (see the description of the triggering of cell senescence in the section "The system telomere-subtelomere-telomerase"). Aging is a physiological phenomenon and, if “disease” is defined as an alteration in the normal conditions, the manifestations of aging cannot be considered diseases, nor can they be “cured”; their treatment would only be the modification of a physiological phenomenon. In contrast, if we modify the definition by considering “disease” as also something that is physiological but causes suffering and/or death, aging can be considered a disease and becomes the object of treatment. However, this modified definition implies an evaluation that is ethical, or more generally non-scientific, because by this definition, a possible treatment that lengthens the life beyond the physiological limits of human would be considered a medical treatment and not a modification of the natural state.
The cases of genetic abnormalities or unhealthy lifestyles that aggravate and/or accelerate aging, or one or more of its manifestations, are different. For example, in a centenarian, some physiological manifestations of aging that may be considered as AD or AMD, etc., are normal, and cannot be considered diseases based on the usual definition. Conversely, if analogous or exacerbated symptoms of the same kind occur at a younger age because of genetic abnormalities or unhealthy lifestyles, they can be considered as diseases in every sense and treated as such. In this case, therefore, the aforementioned ethical problem does not arise. This is important because, in the absence of ethical objections, it will be possible to initiate investigation of strategies for the treatment of such diseases with methods that, when properly developed and tested, may lead to the slowing or even complete control of aging, actions that are complicated by ethical issues.
According to the programmed aging paradigm and taking into account these considerations, a scheme for a tissue or organ in which the main cells show turnover, and its failure (“direct aging”) is presented in Figure 9. A scheme for a tissue or organ in which the main cells do not show turnover while indispensable satellite cells show turnover, and its failure (“indirect aging”) is presented in Figure 10. The medical actions A, B and C are the same as for the programmed and non-programmed aging paradigms. B and C are considered palliative or supplemental both to the medical actions A, which are important, and to the “medical” actions D that are the primary and decisive actions. The term “medical” actions D is used instead of medical actions D, because, as previously stated, they are not medical acts in the strict sense.
However, since the programmed aging paradigm, in its description defined as “telomere theory”, has been considered the working hypothesis of this review, only “medical” actions D will be discussed in the next section.
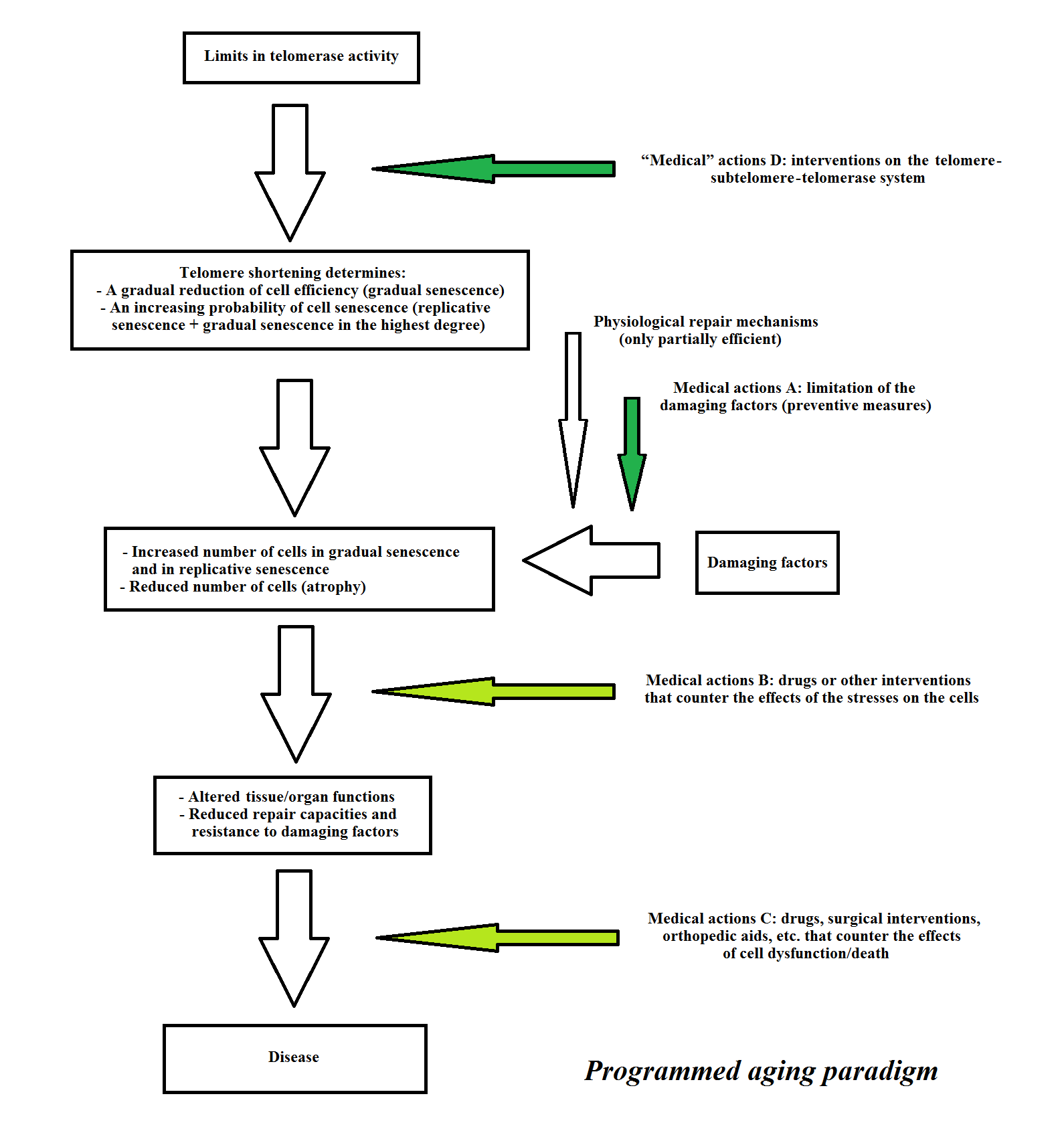
Figure 9 Telomere theory: a scheme for “direct aging”. Medical actions A are important and “medical” actions D are essential, while B and C are palliative or supplemental.
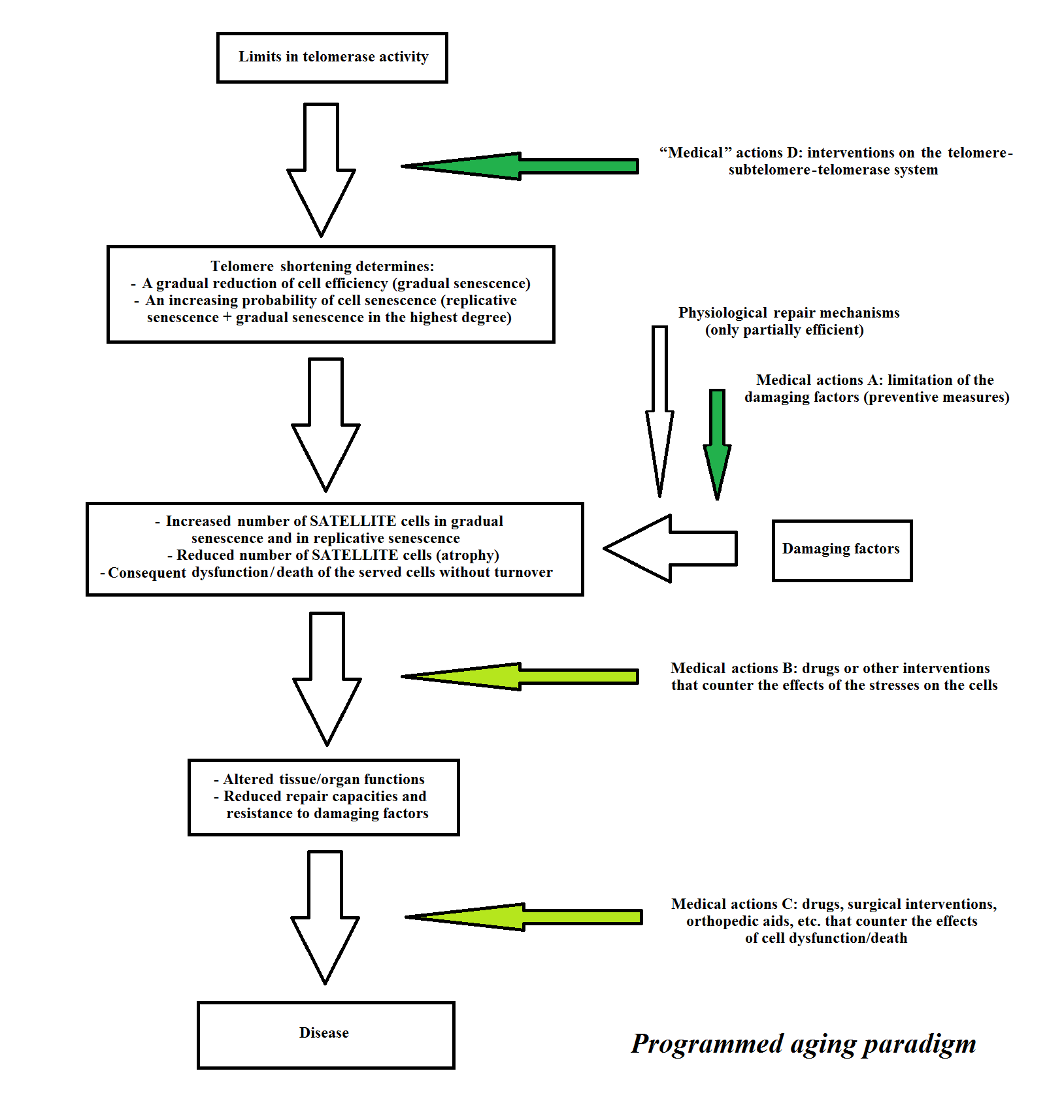
Figure 10 Telomere theory: a scheme for “indirect aging”. Medical actions A are important and “medical” actions D are essential, while B and C are palliative or supplemental.
Potential Treatments for Aging
Three main general approaches are considered here.
(1) Drug-induced Telomerase Activation
It is possible that opportune drugs might stimulate or restore telomerase activity resulting in a reduction in the rate of telomere shortening or even restoration to their initial lengths [216]. Astragalosides have shown a limited capacity in reactivating telomerase [217,218], although these agents are quite expensive and their efficacy is unsatisfactory [216].
A more general objection to this approach is that the existence of drugs that are free from harmful effects and effective in restoration of telomerase activity is not assured in vivo. Research in this direction would also require large investments, and take time without leading to any success. Other approaches with a greater chance of success in a reasonable time-frame should therefore be investigated.
(2) Telomerase Activation by Genetic Methods
One study showed that: “... by using a gene therapy strategy with non-integrative adeno associated virus (AAV), … re-activation of telomerase in adult or old mice results in delayed aging and significant lifespan extension in the absence of increased cancer susceptibility. A single telomerase (TERT) treatment of WT mice with these vectors was sufficient to rescue the age-dependent decline and to delay normal mouse physiological aging ... In this experimental setting, median lifespan was extended by up to 24% in 1-year-old mice, and by 13% in animals of 2 years of age.” [140].
This type of technique (telomerase activation) could be applied in successive experimental phases as follows:
(a) treatment of elderly subjects without reproductive capacity and suffering from severe and/or disabling diseases for which there is no effective cure (e.g., AD, PD and AMD). In this case, there is no ethical problem, as discussed previously, and a successful outcome would pave the way for subsequent phases.
(b) Treatment of elderly subjects without reproductive capacity, and suffering from less disabling diseases or from the aforementioned diseases in a less disabling phase.
(c) Treatment of healthy elderly subjects, also with reproductive capacity, i.e., simple rejuvenation. In this case, the ethical problem exists and such treatment requires comprehensive discussion prior to making a decision.
The theoretical limit of this approach is that, even supposing that periodic treatments are performed, the progressive loss of primary stem cells would not be cancelled by telomerase re-activation (the possible cause of the difference of outcomes in 1- and 2-year-old mice in the study cited previously [107]). Therefore, aging would continue at a reduced pace that would be inversely related to the intensity and frequency of telomerase re-activation treatment.
(3) Telomerase Activation by Genetic Methods Associated with Modification of Subtelomeres and Telomeres
A different method, which would overcome this theoretical limit and amplify the power of telomerase activation, has already been proposed elsewhere [40], so it will only be described here in brief. It is necessary to refer to the original work for details and for the significant ethical issues that the method implies, as it proposes the following genetic changes in germ line cells, i.e., before the period that has been defined as the “reset phase”:
(A) elongation of the telomere by adding further TTAGGG sequences;
(B) insertion of a neutral nucleotide sequence (i.e., without any effect on other parts of the genome) between the subtelomere and the telomere-subtelomere junction (“J”).
The genetic modification obtained by A would allow a greater number of duplications before the telomere reaches a critical size that would not allow further shortening.
The genetic modification obtained by B would allow a greater number of duplications (i.e., a greater relative telomere shortening) before the telomere hood begins to repress the regulatory sequences of the subtelomere.
Actions A and B, combined with periodic telomerase activation treatment, would allow indefinite extension of life, with a minimal or absent progressive loss of primary stem cells, as long as the repressive action of the telomere hood is blocked by telomere lengthening before repression of the regulatory sequences of the subtelomere by the telomere hood is initiated.
These actions are illustrated in Figure 11.
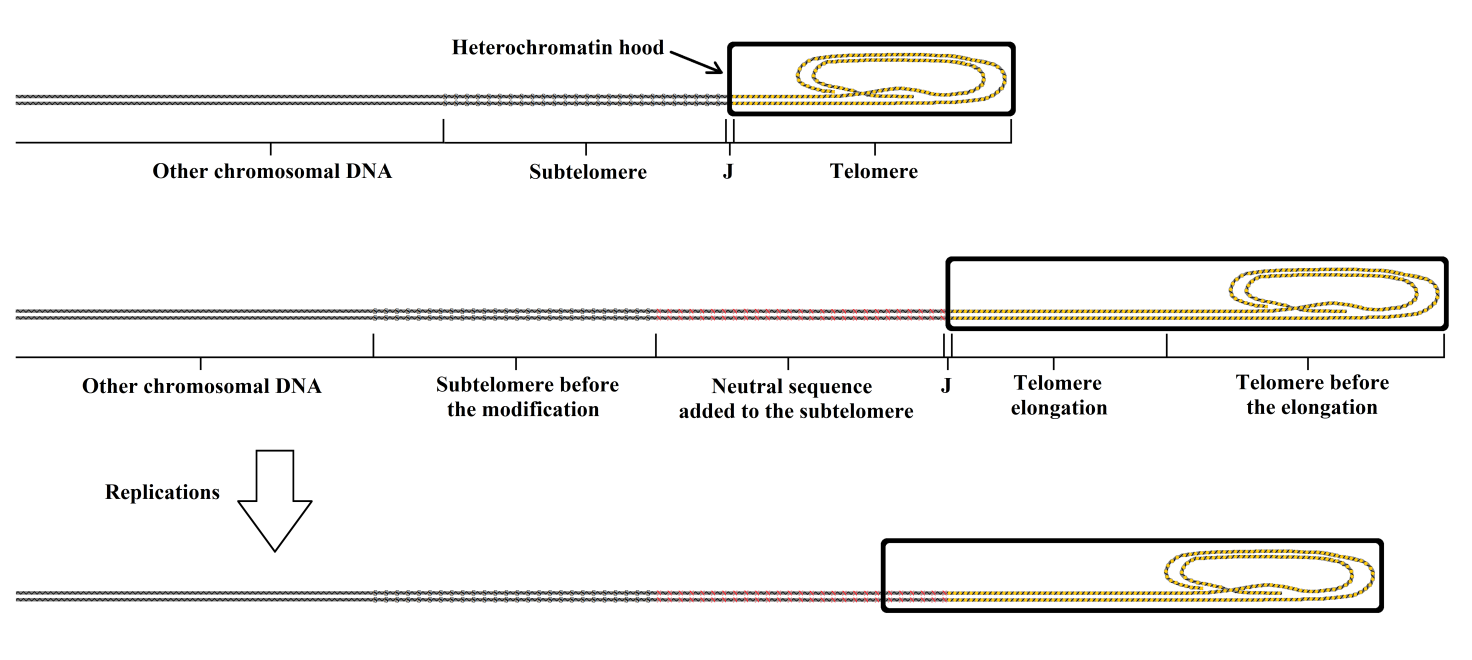
Figure 11 The telomere is elongated before the formation of the telomere hood in the “reset phase”. The greater length of the telomere allows a greater number of duplications before the telomere reaches a critical size that does not allow further shortening. The addition of a neutral sequence between the subtelomere and J allows a greater number of duplications before the subtelomere begins to be repressed by the telomere hood.
Two points must be highlighted. First, the methods proposed here require great reliability and precision, ideally to the level of perfection, of the techniques used for DNA modifications. The best technique that is currently available, and also in economic terms, is the CRISPR-CAS9 (clustered regularly interspaced short palindromic repeat–CRISPR-associated nuclease 9) technique [219,220]. This technique has undergone noteworthy recent improvements [221,222], which have overcome previously known imperfections of the method [223,224]. Second, as explained in a preceding section, the mother lineage cells of yeast (S. cerevisiae) retain the same telomere length with each replication, and with no sliding of the telomere hood over the subtelomere, while there is ERC accumulation over the subtelomere. This progressively represses the subtelomere, so altering cell functions and limiting the number of duplications to a maximum of 25–35 [74]. Yeast could be used as an eukaryotic model to devise simple methods for the insertion of a neutral sequence between the subtelomere and J and to verify if, as expected, this determines an absence of progressive alteration in cell functions and an increase in the maximum number of duplications.
Conclusions
Modern geriatric medicine is unable to act on the primary causes of aging and seeks only to treat the symptoms and complications. It can prolong life, but elderly individuals are disabled more or less severely in the extra years obtained by this approach. In repeating the radical opinion of Abbott on the treatment of AD [210], it is possible to say that the only drugs and medical interventions available to geriatric medicine for senescent patients aim to treat symptoms and are only marginally effective at best. This indicates that a radical change in geriatric medicine is necessary to effectively counter the burden of suffering caused by aging.
Regarding the possibility of such a change, it appears feasible to use the programmed aging paradigm as a working hypothesis in its interpretation defined as “telomere theory”. However, it is essential to abandon the traditional notion that aging is a maladaptive characteristic that is only partially rectifiable [225] and to embrace the new notion that aging is “a specific biological function” [226], which allows a more optimistic approach to possible effective treatment [31,32].
Of course, many will not be convinced by these theories, and perhaps a large majority will consider the explanations of the concepts presented here to be ill-considered and Utopian. Perhaps, the current situation is a “cusp of history”, as defined profoundly by Fossel: “Great ideas pivot on the Janus’d cusp of history: looking forward they are obviously foolish, looking backward they appear foolishly obvious. We are doubly blind” [39]. However, if these theories are substantiated, they would constitute a scientific revolution or a paradigm-shift in the sense defined by Kuhn [16].
Furthermore, if the paradigm-shift offers the potential to apply techniques effectively to increase the lifespan further or indefinitely, this will represent a seismic change in our civilization [32]. Thus, it would not be an exaggeration to consider this as a universal and momentous revolution. As in every revolution, even in the best possible, not everything will change for the better. The greatest danger is indicated in ancient Greek wisdom. The gods were conceived by the ancient Greeks as immortal beings and the worst sin was to be guilty of “ύβρις”, which is the aspiration to reach god-like status with impious unforgivable arrogance and pride. Perhaps the wisest choice would be abstain from changing anything, but unknown lands are irresistible to humans. However, the choices presented by the possible options do not fall within the scope of this review or science overall.
References
- Libertini G. An adaptive theory of the increasing mortality with increasing chronological age in populations in the wild. J Theor Biol. 1988;132:145-62. [CrossRef]
- Holmes DJ, Austad SN. Birds as animal models for the comparative biology of aging: a prospectus. J Gerontol A Biol Sci. 1995;50(2):B59-66. [CrossRef]
- Rose MR. Evolutionary biology of aging. New York: Oxford University Press; 1991; p. 20, 21. [CrossRef]
- Kirkwood TB, Austad SN. Why do we age? Nature. 2000;408:233-8. [CrossRef]
- Flatt T, Schmidt PS. Integrating evolutionary and molecular genetics of aging. Biochim Biophys Acta. 2009;1790(10):951-62. [CrossRef]
- Williams GC. Pleiotropy, natural selection and the evolution of senescence. Evolution. 1957;11:398-411. [CrossRef]
- Nussey DH, Froy H, Lemaitre JF, Gaillard JM, Austad SN. Senescence in natural populations of animals: widespread evidence and its implications for bio-gerontology. Ageing Res Rev. 2013;12:214-25. [CrossRef]
- Hill K, Hurtado AM. Ache life history. New York: Aldine De Gruyter; 1996.
- Finch CE. Longevity, senescence, and the genome. Chicago: The University of Chicago Press; 1990.
- Ricklefs RE. Evolutionary theories of aging: confirmation of a fundamental prediction, with implications for the genetic basis and evolution of life span. Am Nat. 1998;152(1):24-44. [CrossRef]
- Comfort A. The biology of senescence. New York: Elsevier North Holland; 1979.
- Medvedev ZA. An attempt at a rational classification of theories of ageing. Biol Rev Camb Philos Soc. 1990;65(3):375-98. [CrossRef]
- Libertini G. Phylogeny of Aging and Related Phenoptotic Phenomena. Biochem (Mosc). 2015;80(12):1529-46. [CrossRef]
- Libertini G. Empirical evidence for various evolutionary hypotheses on species demonstrating increasing mortality with increasing chronological age in the wild. Scientific World Journal. 2008;8:182-93. [CrossRef]
- Libertini G. Non-programmed versus programmed aging paradigm. Curr Aging Sci. 2015;8(1):56-68. [CrossRef]
- Kuhn TS. The Structure of Scientific Revolutions. Chicago: The University of Chicago Press; 1962.
- Medawar PB. An unsolved problem in biology. London: H. K. Lewis; 1952. Reprinted in: Medawar PB. The uniqueness of the individual. London: Methuen; 1957. [CrossRef]
- Hamilton WD. The moulding of senescence by natural selection. J Theor Biol. 1966;12(1):12-45. [CrossRef]
- Kirkwood TB. Evolution of ageing. Nature. 1977;270:301-4. [CrossRef]
- Kirkwood TB, Holliday R. The evolution of ageing and longevity. Proc R Soc Lond B Biol Sci. 1979;205:531-46. [CrossRef]
- ICD-9-CM, 2016. Available: http://www.cdc.gov/nchs/icd/icd9cm.htm.
- ICD-10, 2016. Available: http://www.who.int/classifications/apps/icd/icd10online/.
- World Ranking Total Deaths, 2014. Available: http://www.worldlifeexpectancy.com/worldrankings-total-deaths. See also: http://www.worldlifeexpectancy.com/sitemap.
- Skulachev VP. Mitochondrial physiology and pathology; concepts of programmed death of organelles, cells and organisms. Mol Aspects Med. 1999;20(3):139-84. [CrossRef]
- Libertini G. Classification of phenoptotic phenomena. Biochem (Mosc). 2012;77(7):707-15. [CrossRef]
- Pianka ER. On r- and K-selection. Amer Natur. 1970;104:592-7. [CrossRef]
- Travis JM. The evolution of programmed death in a spatially structured population. J Gerontol A Biol Sci Med Sci. 2004, 59(4):301-5. [CrossRef]
- Martins AC. Change and aging senescence as an adaptation. PLoS One. 2011;6(9):e24328. [CrossRef]
- Mitteldorf J, Martins AC. Programmed life span in the context of evolvability. Am Nat. 2014;184(3):289-302. [CrossRef]
- Lenart P, Bienertová-Vašků J. Keeping up with the Red Queen: the pace of aging as an adaptation. Biogerontology. 2016;DOI 10.1007/s10522-016-9674-4. [CrossRef]
- Libertini G. Prospects of a Longer Life Span beyond the Beneficial Effects of a Healthy Lifestyle. In: Bentely JV, Keller MA (eds.). Handbook on longevity: genetics, diet and disease. New York: Nova Science Publ. Inc.; 2009; pp. 35-96.
- Libertini G. The Role of Telomere-Telomerase System in Age-Related Fitness Decline, a Tameable Process. In: Mancini L (ed.). Telomeres: Function, Shortening and Lengthening. New York: Nova Science Publ. Inc.; 2009; pp. 77-132.
- Libertini G. The programmed aging paradigm: how we get old. Biochem (Mosc). 2014;79(10):1004-16. [CrossRef]
- Skulachev VP. Phenoptosis: programmed death of an organism. Biochem (Mosc). 1999;64(12):1418-26.
- Olovnikov AM. The redusome hypothesis of aging and the control of biological time during individual development. Biochem (Mosc). 2003;68(1):2-33. [CrossRef]
- Olovnikov AM. Chronographic theory of development, aging, and origin of cancer: role of chronomeres and printomeres. Curr Aging Sci. 2015;8(1):76-88. [CrossRef]
- Skulachev VP, Longo VD. Aging as a mitochondria-mediated atavistic program: can aging be switched off? Ann N Y Acad Sci. 2005;1057:145-64. [CrossRef]
- Goldsmith TC. Aging, evolvability, and the individual benefit requirement; medical implications of aging theory controversies. J Theor Biol. 2008;252(4):764-8. [CrossRef]
- Fossel MB. Cells, Aging and Human Disease. New York: Oxford University Press; 2004.
- Libertini G, Ferrara N. Possible interventions to modify aging. Biochem (Mosc). 2016, 81(12):1413-28. [CrossRef]
- Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, et al. A highly conserved repetitive DNA sequence (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci USA. 1988;85(18):6622-6. [CrossRef]
- Blackburn EH. Structure and function of telomeres. Nature. 1991;350:569-73. [CrossRef]
- Mefford HC, Trask BJ. The complex structure and dynamic evolution of human subtelomeres. Nat Rev Genet. 2002;3(2):91-102. [CrossRef]
- Torres GA, Gong Z, Iovene M, Hirsch CD, Buell CR, Bryan GJ, et al. Organization and evolution of subtelomeric satellite repeats in the potato genome. G3 (Bethesda). 2011;1(2):85-92. [CrossRef]
- Olovnikov AM. Principle of marginotomy in template synthesis of polynucleotides [in Russian]. Dokl Akad Nauk SSSR. 1971;201(6):1496-9. English version: Doklady Biochem. 1971;201:394-7.
- Watson JD. Origin of concatemeric T7 DNA. Nat New Biol. 1972;239(94):197-201. [CrossRef]
- Olovnikov AM. A theory of marginotomy: The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the problem. J Theor Biol. 1973;41(1):181-90. [CrossRef]
- Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43(2 Pt 1):405-13. [CrossRef]
- van Steensel B, de Lange T. Control of telomere length by the human telomeric protein TRF1. Nature. 1997;385:740-3. [CrossRef]
- Takubo K, Aida J, Izumiyama-Shimomura N, Ishikawa N, Sawabe M, Kurabayashi R, et al. Changes of telomere length with aging. Geriatr Gerontol Int. 2010;10:S197-206. [CrossRef]
- Haussmann MF, Winkler DW, Vleck CM. Longer telomeres associated with higher survival in birds. Biol Lett. 2005;1(2):212-4. [CrossRef]
- Pauliny A, Wagner RH, Augustin J, Szép T, Blomqvist D. Age-independent telomere length predicts fitness in two bird species. Mol Ecol. 2006;15(6):1681-7. [CrossRef]
- Bize P, Criscuolo F, Metcalfe NB, Nasir L, Monaghan P. Telomere dynamics rather than age predict life expectancy in the wild. Proc Biol Sci. 2009;276:1679-83. [CrossRef]
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585-621. [CrossRef]
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614-36. [CrossRef]
- Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6(3):331-43. [CrossRef]
- Bierman EL. The effect of donor age on the in vitro life span of cultured human arterial smooth-muscle cells. In Vitro. 1978;14(11):951-5. [CrossRef]
- Tassin J, Malaise E, Courtois Y. Human lens cells have an in vitro proliferative capacity inversely proportional to the donor age. Exp Cell Res. 1979;123(2):388-92. [CrossRef]
- Schneider EL, Mitsui Y. The relationship between in vitro cellular aging and in vivo human age. Proc Natl Acad Sci USA. 1976;73(10):3584-8. [CrossRef]
- Martin GM, Sprague CA, Epstein CJ. Replicative life-span of cultivated human cells. Effects of donor’s age, tissue, and genotype. Lab Invest. 1970;23(1):86-92.
- Röhme D. Evidence for a relationship between longevity of mammalian species and life spans of normal fibroblasts in vitro and erythrocytes in vivo. Proc Natl Acad Sci USA. 1981;78(8):5009-13. [CrossRef]
- Pontén J, Stein WD, Shall S. A quantitative analysis of the aging of human glial cells in culture. J Cell Phys. 1983;117(3):342-52. [CrossRef]
- Jones RB, Whitney RG, Smith JR. Intramitotic variation in proliferative potential: stochastic events in cellular aging. Mech Ageing Dev. 1985;29(2):143-9. [CrossRef]
- Blackburn EH. Telomere states and cell fates. Nature. 2000;408:53-6. [CrossRef]
- Ben-Porath I, Weinberg R. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37(5):961-76. [CrossRef]
- Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell. 1990;63(4):751-62. [CrossRef]
- Robin JD, Ludlow AT, Batten K, Magdinier F, Stadler G, Wagner KR et al. Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances. Genes Dev. 2014;28(22):2464-76. [CrossRef]
- Slijepcevic P, Hande MP. Chinese hamster telomeres are comparable in size to mouse telomeres. Cytogenet Cell Genet. 1999;85(3-4):196-9. [CrossRef]
- Gorbunova V, Bozzella MJ, Seluanov A. Rodents for comparative aging studies: from mice to beavers. Age (Dordr). 2008;30(2-3):111-9. [CrossRef]
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91(1):25-34. [CrossRef]
- Herrera E, Samper E, Martín-Caballero J, Flores JM, Lee HW, Blasco MA. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres, EMBO J. 1999;18(11):2950-60. [CrossRef]
- Lee HW, Blasco MA, Gottlieb GJ, Horner JW 2nd, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569-74. [CrossRef]
- Prowse KR, Greider CW. Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc Natl Acad Sci USA. 1995;92(11):4818-22. [CrossRef]
- Jazwinski SM. The genetics of aging in the yeast Saccharomyces cerevisiae. Genetica. 1993(13);91:35-51. [CrossRef]
- Laun P, Pichova A, Madeo F, Fuchs J, Ellinger A, Kohlwein S, et al. Aged mother cells of Saccharomyces cerevisiae show markers of oxidative stress and apoptosis. Mol Microbiol. 2001;39(5):1166-73. [CrossRef]
- Herker E, Jungwirth H, Lehmann KA, Maldener C, Fröhlich KU, Wissing S, et al. Chronological aging leads to apoptosis in yeast. J Cell Biol. 2004;164(4):501-7. [CrossRef]
- Lesur I, Campbell JL. The transcriptome of prematurely aging yeast cells is similar to that of telomerase-deficient cells. Mol Biol Cell. 2004;15(3):1297-312. [CrossRef]
- Büttner S, Eisenberg T, Herker E, Carmona-Gutierrez D, Kroemer G, Madeo F. Why yeast cells can undergo apoptosis: death in times of peace, love, and war. J Cell Biol. 2006;175(4):521-5. [CrossRef]
- Fabrizio P, Longo VD. Chronological aging-induced apoptosis in yeast. Biochim Biophys Acta. 2008;1783(7):1280-5. [CrossRef]
- Laun P, Bruschi CV, Dickinson JR, Rinnerthaler M, Heeren G, Schwimbersky R, et al. Yeast mother cell-specific ageing, genetic (in)stability, and the somatic mutation theory of ageing. Nucleic Acids Res. 2007;35(22):7514-26. [CrossRef]
- D’Mello NP, Jazwinski SM. Telomere length constancy during aging of Saccharomyces cerevisiae. J Bacteriol. 1991;173(21):6709-13. [CrossRef]
- Smeal T, Claus J, Kennedy B, Cole F, Guarente L. Loss of transcriptional silencing causes sterility in old mother cells of Saccharomyces cerevisiae. Cell. 1996;84(4):633-42. [CrossRef]
- Maringele L, Lydall D. Telomerase- and recombination-independent immortalization of budding yeast. Genes Dev. 2004;18(21):2663-75. [CrossRef]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell. 1997;91(7):1033-42. [CrossRef]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239-57. [CrossRef]
- Kaeberlein M, Burtner CR, Kennedy BK. Recent developments in yeast aging. PLoS Genetics. 2007;3(5):e84. [CrossRef]
- Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251-306. [CrossRef]
- Lynch MP, Nawaz S, Gerschenson LE. Evidence for soluble factors regulating cell death and cell proliferation in primary cultures of rabbit endometrial cells grown on collagen. Proc Natl Acad Sci USA. 1986;83(13):4784-8. [CrossRef]
- Medh RD, Thompson EB. Hormonal regulation of physiological cell turnover and apoptosis. Cell Tissue Res. 2000;301(1):101-24. [CrossRef]
- Benedetti A, Jezequel AM, Orlandi F. A quantitative evaluation of apoptotic bodies in rat liver. Liver. 1988;8(3):172-7. [CrossRef]
- Dremier S, Golstein J, Mosselmans R, Dumont JE, Galand P, Robaye B. Apoptosis in dog thyroid cells. Biochem Biophys Res Comm. 1994;200(1):52-8. [CrossRef]
- Finegood DT, Scaglia L, Bonner-Weir S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes. 1995;44(3):249-56. [CrossRef]
- Prins JB, O’Rahilly S. Regulation of adipose cell number in man. Clin Sci (Lond). 1997;92(1):3-11. [CrossRef]
- Migheli A, Mongini T, Doriguzzi C, Chiadò-Piat L, Piva R, Ugo I, Palmucci L. Muscle apoptosis in humans occurs in normal and denervated muscle, but not in myotonic dystrophy, dystrophinopathies or inflammatory disease. Neurogenetics. 1997;1(2):81-7. [CrossRef]
- Pollack M, Leeuwenburgh C. Apoptosis and aging: role of the mitochondria. J Gerontol A Biol Sci Med Sci. 2001;56(11):B475-82. [CrossRef]
- Spelsberg TC, Subramaniam M, Riggs BL, Khosla S. The actions and interactions of sex steroids and growth factors/cytokines on the skeleton. Mol Endocrinol. 1999;13(6):819-28. [CrossRef]
- Héraud F, Héraud A, Harmand MF. Apoptosis in normal and osteoarthritic human articular cartilage. Ann Rheum Dis. 2000;59(12):959-65. [CrossRef]
- Cardani R, Zavanella T. Age-related cell proliferation and apoptosis in the kidney of male Fischer 344 rats with observations on a spontaneous tubular cell adenoma. Toxicol Pathol. 2000;28(6):802-6. [CrossRef]
- Harada K, Iwata M, Kono N, Koda W, Shimonishi T, Nakanuma Y. Distribution of apoptotic cells and expression of apoptosis-related proteins along the intrahepatic biliary tree in normal and non-biliary diseased liver. Histopathology. 2000;37(4):347-54. [CrossRef]
- Xia SJ, Xu CX, Tang XD, Wang WZ, Du DL. Apoptosis and hormonal milieu in ductal system of normal prostate and benign prostatic hyperplasia. Asian J Androl. 2001;3(2):131-4.
- Sutherland LM, Edwards YS, Murray AW. Alveolar type II cell apoptosis. Comp Biochem Physiol. 2001;129(1):267-85. [CrossRef]
- Richardson RB, Allan DS, Le Y. Greater organ involution in highly proliferative tissues associated with the early onset and acceleration of ageing in humans. Exp Geront. 2014;55:80-91. [CrossRef]
- Alberts B, Bray D, Hopkin K, Johnson A, Lewis J, Raff M, Roberts K, Walter P (eds.). Essential Cell Biology. 4th ed. New York: Garland Science; 2014. [CrossRef]
- Anversa P, Kajstura J, Leri A, Bolli R. Life and death of cardiac stem cells. Circulation. 2006;113(11):1451-63. [CrossRef]
- Reed JC. Dysregulation of apoptosis in cancer. J Clin Oncol. 1999;17(9):2941-53. [CrossRef]
- DePinho RA. The age of cancer. Nature. 2000;408:248-54. [CrossRef]
- Bernardes de Jesus B, Blasco MA. Potential of telomerase activation in extending health span and longevity. Curr Opin Cell Biol. 2012;24(6):739-43. [CrossRef]
- Horner PJ, Gage FH. Regenerating the damaged central nervous system. Nature. 2000;407:963-70. [CrossRef]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132(4):645-60. [CrossRef]
- Williams RW, Herrup K. The control of neuron number. Annu Rev Neurosc. 1988;11(1):423-53. [CrossRef]
- Bermingham-McDonogh O, Reh TA. Regulated reprogramming in the regeneration of sensory receptor cells. Neuron. 2011;71(3):389-405. [CrossRef]
- Maier EC, Saxena A, Alsina B, Bronner ME, Whitfield TT. Sensational placodes: neurogenesis in the otic and olfactory systems. Dev Biol. 2014;389(1):50-67. [CrossRef]
- Fine SL, Berger JW, Maguire MG, Ho AC. Age-related macular degeneration. N Engl J Med. 2000;342(7):483-92. [CrossRef]
- Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N Engl J Med. 2008;358(24):2606-17. [CrossRef]
- Sparrow JR. Therapy for macular degeneration: insights from acne. Proc Natl Acad Sci USA. 2003;100(8):4353-4. [CrossRef]
- Berger JM, Fine SL, Maguire MG. Age-related macular degeneration. USA: Mosby; 1999.
- Flanary B. Telomeres: Function, Shortening, and Lengthening. In: Mancini L (ed.). Telomeres: Function, Shortening and Lengthening. New York: Nova Science Publ. Inc., New York; 2009; pp. 379-386.
- Libertini G, Ferrara N. Aging of perennial cells and organ parts according to the programmed aging paradigm. Age (Dordr). 2016;38(2):35. doi:10.1007/s11357-016-9895-0. [CrossRef]
- Martin GM, Oshima J. Lessons from human progeroid syndromes. Nature. 2000;408:263-6. [CrossRef]
- Marchesini G, Bua V, Brunori A, Bianchi G, Pisi P, Fabbri A et al. Galactose elimination capacity and liver volume in aging man. Hepatology. 1988;8(5):1079-83. [CrossRef]
- Wynne HA, Cope, LH, Mutch E, Rawlins MD, Woodhouse KW, James OF. The effect of age upon liver volume and apparent liver blood flow in healthy man. Hepatology. 1989;9(2):297-301. [CrossRef]
- David H, Reinke P. Liver morphology with aging. In: Bianchi L, Holt P, James OFW, Butker RN (eds). Aging in Liver and Gastrointestinal tracts. Lancaster (UK): MTP Press; 1988; pp. 143-59.
- Watanabe T, Tanaka, Y. Age-related alterations in the size of human epatocytes. A study of mononuclear and binucleate cells. Virchow Arch. 1982;39(1):9-20. [CrossRef]
- Leri A, Barlucchi L, Limana F, Deptala A, Darzynkiewicz Z, Hintze TH, et al. Telomerase expression and activity are coupled with myocyte proliferation and preservation of telomeric length in the failing heart. Proc Natl Acad Sci USA. 2001;98(15):8626-31. [CrossRef]
- Urbanek K, Quaini F, Tasca G, Torella D, Castaldo C, Nadal-Ginard B, et al. Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy. Proc Natl Acad Sci USA. 2003;100(18):10440-5. [CrossRef]
- Chimenti C, Kajstura J, Torella D, Urbanek K, Heleniak H, Colussi C, et al. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res. 2003;93(7):604-13. [CrossRef]
- Olivetti G, Melissari M, Capasso JM, Anversa P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res. 1991;68(6):1560-8. [CrossRef]
- Aronow WS. Effects of Aging on the Heart. In: Tallis RC, Fillit HM, Brocklehurst JC. (eds). Brocklehurst’s Textbook of Geriatric Medicine and Gerontology. 5th ed. New York: Churchill Livingstone; 1998; pp. 255-62.
- Griffiths CEM. Aging of the Skin. In: Tallis RC, Fillit HM, Brocklehurst JC. (eds). Brocklehurst’s Textbook of Geriatric Medicine and Gerontology. 5th ed. New York: Churchill Livingstone; 1998; pp. 1293-8.
- Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348(7):593-600. [CrossRef]
- Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353(10):999-1007. [CrossRef]
- Tallis RC, Fillit HM, Brocklehurst JC. (eds). Brocklehurst’s Textbook of Geriatric Medicine and Gerontology. 5th ed. New York: Churchill Livingstone; 1998.
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu C, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349-52. [CrossRef]
- Counter CM, Hahn WC, Wei W, Caddle SD, Beijersbergen RL, Lansdorp PM, et al. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci USA. 1998;95(25):14723-8. [CrossRef]
- Vaziri H. Extension of life span in normal human cells by telomerase activation: a revolution in cultural senescence. J Anti-Aging Med. 1998;1(2):125-30. [CrossRef]
- Vaziri H, Benchimol S. Reconstitution of telomerase activity in human normal cells leads to elongation of telomeres and extended replicative life span. Curr Biol. 1998;8(5):279-82. [CrossRef]
- de Lange T, Jacks, T. For better or worse? Telomerase inhibition and cancer. Cell. 1999;98(3):273-5. [CrossRef]
- Funk WD, Wang CK, Shelton DN, Harley CB, Pagon GD, Hoeffler WK. Telomerase expression restores dermal integrity to in vitro-aged fibroblasts in a reconstituted skin model. Exp Cell Res. 2000;258(2):270-8. [CrossRef]
- Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011;469:102-6. [CrossRef]
- Bernardes de Jesus B, Vera E, Schneeberger K, Tejera AM, Ayuso E, Bosch F, Blasco MA. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med. 2012;4(8):691-704. [CrossRef]
- Kirkwood TB, Melov S. On the programmed/non-programmed nature of ageing within the life history. Curr Biol. 2011;21(18):R701-7. [CrossRef]
- Bredesen DE. The non-existent aging program: how does it work? Aging Cell. 2004;3(5):255-9. [CrossRef]
- Blagosklonny MV. Aging is not programmed: genetic pseudo-program is a shadow of developmental growth. Cell Cycle. 2013;12(24):3736-42. [CrossRef]
- Hayflick L. Unlike Aging, Longevity Is Sexually Determined. In: Bengtson VL, Settersten RA Jr (eds). Handbook of Theories of Aging. 3rd ed. New York: Springer; 2016.
- Campisi J. The biology of replicative senescence. Eur J Cancer. 1997;33(5):703-9. [CrossRef]
- Wright WE, Shay JW. Telomere biology in aging and cancer. J Am Geriatr Soc. 2005;53(9):S292-4. [CrossRef]
- Campisi J. Cancer, aging and cellular senescence. In Vivo. 2000;14(1):183-8. [CrossRef]
- Libertini G. Evidence for aging theories from the study of a hunter–gatherer people (Ache of Paraguay). Biochem (Mosc). 2013;78(9):1023-32. [CrossRef]
- Mitteldorf J. Telomere biology: Cancer firewall or aging clock? Biochem (Mosc). 2013;78(9):1054-60. [CrossRef]
- Fabrizio P, Longo VD. The chronological life span of Saccharomyces cerevisiae. Methods Mol Biol. 2007;371:89-95. [CrossRef]
- Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 2014;15(11):1139-53. [CrossRef]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184-9. [CrossRef]
- Rosen P. Aging of the immune system. Med Hypotheses. 1985;18(2):157-61. [CrossRef]
- Klapper W, Heidorn K, Kühne K, Parwaresch R, Krupp G. Telomerase activity in ‘immortal’ fish. FEBS Letters. 1998;434(3):409-12. [CrossRef]
- Klapper W, Kühne K, Singh KK, Heidorn K, Parwaresch R, Krupp, G. Longevity of lobsters is linked to ubiquitous telomerase expression. FEBS Letters. 1998;439(1-2):143-6. [CrossRef]
- Artandi SE. Telomere shortening and cell fates in mouse models of neoplasia. Trends Mol Med. 2002;8(1):44-7. [CrossRef]
- Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010;31(1):9-18. [CrossRef]
- González-Suárez E, Samper E, Ramirez A, Flores JM, Martín-Caballero J, Jorcano JL, Blasco MA. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. EMBO J. 2001;20(11):2619-30. [CrossRef]
- González-Suárez E, Geserick C, Flores JM, Blasco MA. Antagonistic effects of telomerase on cancer and aging in K5-mTert transgenic mice. Oncogene. 2005;24(13):2256-70. [CrossRef]
- Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol. 2000;110(4):768-79. [CrossRef]
- Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413:432-5. [CrossRef]
- Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402:551-5. [CrossRef]
- Marciniak R, Guarente L. Human genetics. Testing telomerase. Nature. 2001;413:370-2. [CrossRef]
- Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, et al. (1996) Positional cloning of the Werner's syndrome gene. Science. 1996;272:258-62. [CrossRef]
- Fukuchi K, Martin GM, Monnat RJ Jr. Mutator phenotype of Werner syndrome is characterized by extensive deletions. Proc Natl Acad Sci USA. 1989;86(20):5893-7. Published erratum appears in Proc Natl Acad Sci USA. 1989;86(20):7994. [CrossRef]
- Wilson PW, Castelli WP, Kannel WB. Coronary risk prediction in adults (the Framingham Heart Study). Am J Cardiol. 1987;59(14):91-4G. [Erratum, Am. J. Cardiol. 1987;60(13):A11.] [CrossRef]
- Roerecke M, Rehm J. Alcohol consumption, drinking patterns, and ischemic heart disease: a narrative review of meta-analyses and a systematic review and meta-analysis of the impact of heavy drinking occasions on risk for moderate drinkers. BMC Med. 2014;12:182. doi:10.1186/s12916-014-0182-6. [CrossRef]
- Gardner JD, Mouton AJ. Alcohol effects on cardiac function. Compr Physiol. 2015;(2):791-802. [CrossRef]
- de Gaetano G, Costanzo S, Di Castelnuovo A, Badimon L, Bejko D, Alkerwi A, et al. Effects of moderate beer consumption on health and disease: A consensus document. Nutr Metab Cardiovasc Dis. 2016;26(6):443-67. [CrossRef]
- Walter DH, Rittig K, Bahlmann FH, Kirchmair R, Silver M, Murayama T. et al. Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation. 2002;105(25):3017-24. [CrossRef]
- Su JB. Vascular endothelial dysfunction and pharmacological treatment. World J Cardiol. 2015;7(11):719-41. [CrossRef]
- Rudnicka AR, Jarrar Z, Wormald R, Cook DG, Fletcher A, Owen CG. Age and gender variations in age-related macular degeneration prevalence in populations of European ancestry: a meta-analysis. Ophthalmology. 2012;119(3):571-80. [CrossRef]
- Klein R, Deng Y, Klein BE, Hyman L, Seddon J, Frank RN, et al. Cardiovascular disease, its risk factors and treatment, and age-related macular degeneration: Women's Health Initiative Sight Exam ancillary study. Am J Ophthalmol. 2007;143(3):473-83. [CrossRef]
- Mares JA, Voland RP, Sondel SA, Millen AE, Larowe T, Moeller SM, et al. Healthy lifestyles related to subsequent prevalence of age-related macular degeneration. Arch Ophthalmol. 2011;129(4):470-80. [CrossRef]
- Armstrong RA, Mousavi M. Overview of risk factors for age-related macular degeneration (AMD). J Stem Cells. 2015;10(3):171-91.
- Klein R, Cruickshanks KJ, Nash SD, Krantz EM, Nieto FJ, Huang GH, et al. The prevalence of age-related macular degeneration and associated risk factors. Arch Ophthalmol. 2010;128(6):750-8. [CrossRef]
- Gehlbach P, Li T, Hatef E. Statins for age-related macular degeneration. Cochrane Database Syst Rev. 2015;(2):CD006927. doi:10.1002/14651858.CD006927.pub4. [CrossRef]
- Gorelick PB. Risk factors for vascular dementia and Alzheimer disease. Stroke. 2004;35(11 Suppl 1):2620-2. [CrossRef]
- Rosendorff C, Beeri MS, Silverman JM. Cardiovascular risk factors for Alzheimer’s disease. Am J Geriatr Cardiol. 2007;16(3):143-9. [CrossRef]
- Baglietto-Vargas D, Shi J, Yaeger DM, Ager R, LaFerla FM. Diabetes and Alzheimer’s disease crosstalk. Neurosci Biobehav Rev. 2016;64:272-87. [CrossRef]
- Rani V, Deshmukh R, Jaswal P, Kumar P, Bariwal J. Alzheimer’s disease: Is this a brain specific diabetic condition? Physiol Behav. 2016, 164(Pt A):259-67. [CrossRef]
- Saedi E, Gheini MR, Faiz F, Arami MA. Diabetes mellitus and cognitive impairments. World J Diabetes. 2016;7(17):412-22. [CrossRef]
- Vicente Miranda H, El-Agnaf OM, Outeiro TF. Glycation in Parkinson’s disease and Alzheimer’s disease. Mov Disord. 2016;31(6):782-90. [CrossRef]
- Wanamaker BL, Swiger KJ, Blumenthal RS, Martin SS. Cholesterol, statins, and dementia: what the cardiologist should know. Clin Cardiol. 2015;38(4):243-50. [CrossRef]
- Qiu C, Winblad B, Fratiglioni L. The age-dependent relation of blood pressure to cognitive function and dementia. Lancet Neurol. 2005;4(8):487-99. [CrossRef]
- Michel JP. Is it possible to delay or prevent age-related cognitive decline? Korean J Fam Med. 2016;37(5):263-6. [CrossRef]
- Durazzo TC, Mattsson N, Weiner MW. Smoking and increased Alzheimer’s disease risk: a review of potential mechanisms. Alzheimers Dement. 2014;10(3 Suppl):S122-45. [CrossRef]
- Campdelacreu J. Parkinson disease and Alzheimer disease: environmental risk factors. Article in English, Spanish. Neurologia. 2014;29(9):541-9. [CrossRef]
- Ilomaki J, Jokanovic N, Tan EC, Lonnroos E. Alcohol consumption, dementia and cognitive decline: an overview of systematic reviews. Curr Clin Pharmacol. 2015;10(3):204-12. [CrossRef]
- Vogel T, Benetos A, Verreault R, Kaltenbach G, Kiesmann M, Berthel M. [Risk factors for Alzheimer: towards prevention?]. [Article in French]. Presse Med. 2006;35(9 Pt 2):1309-16. [CrossRef]
- Ellul J, Archer N, Foy CM, Poppe M, Boothby H, Nicholas H, et al. The effects of commonly prescribed drugs in patients with Alzheimer’s disease on the rate of deterioration. J Neurol Neurosurg Psychiatry. 2007;78(3):233-9. [CrossRef]
- Yasar S, Schuchman M, Peters J, Anstey KJ, Carlson MC, Peters R. Relationship between antihypertensive medications and cognitive impairment: Part I. Review of human studies and clinical trials. Curr Hypertens Rep. 2016, 18(8):67. doi:10.1007/s11906-016-0674-1. [CrossRef]
- de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5(6):525-35. [CrossRef]
- Pringsheim T, Jette N, Frolkis A, Steeves TD. The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov Disord. 2014;29(13):1583-90. [CrossRef]
- Hu G, Jousilahti P, Bidel S, Antikainen R, Tuomilehto J. Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care. 2007, 30(4):842-7. [CrossRef]
- Tomlinson DR, Gardiner NJ. Glucose neurotoxicity. Nat Rev Neurosci. 2008;9(1):36-45. [CrossRef]
- Zhang P, Tian B. Metabolic syndrome: an important risk factor for Parkinson’s disease. Oxid Med Cell Longev. 2014;2014:729194. doi:10.1155/2014/729194. [CrossRef]
- Abbott RD, Ross GW, White LR, Nelson JS, Masaki KH, Tanner CM, et al. Midlife adiposity and the future risk of Parkinson's disease. Neurology. 2002;59(7):1051-7. [CrossRef]
- Hu G, Jousilahti P, Nissinen A, Antikainen R, Kivipelto M, Tuomilehto J. Body mass index and the risk of Parkinson disease. Neurology. 2006;67(11):1955-9. [CrossRef]
- Li J, Cesari M, Liu F, Dong B, Vellas B. Effects of diabetes mellitus on cognitive decline in patients with Alzheimer disease: a systematic review. Can J Diabetes. 2016;41(1):114-9. [CrossRef]
- Ishihara L, Brayne C. A systematic review of nutritional risk factors of Parkinson’s disease. Nutr Res Rev. 2005;18(2):259-82. [CrossRef]
- Eriksson AK, Löfving S, Callaghan RC, Allebeck P. Alcohol use disorders and risk of Parkinson’s disease: findings from a Swedish national cohort study 1972-2008. BMC Neurol. 2013;13:190. doi:10.1186/1471-2377-13-190. [CrossRef]
- Gao X, Simon KC, Schwarzschild MA, Ascherio A. A prospective study of statin use and risk of Parkinson disease. Arch Neurol. 2012;69(3):380-4. [CrossRef]
- Friedman B, Lahad A, Dresner Y, Vinker S. Long-term statin use and the risk of Parkinson’s disease. Am J Manag Care. 2013;19(8):626-32.
- Undela K, Gudala K, Malla S, Bansal D. Statin use and risk of Parkinson’s disease: a metaanalysis of observational studies. J Neurol. 2013;260(1):158-65. [CrossRef]
- Sheng Z, Jia X, Kang M. Statin use and risk of Parkinson’s disease: A meta-analysis. Behav Brain Res. 2016;309:29-34. [CrossRef]
- Lopez-Real A, Rey P, Soto-Otero R, Mendez-Alvarez E, Labandeira-Garcia JL. Angiotensinconverting enzyme inhibition reduces oxidative stress and protects dopaminergic neurons in a 6-hydroxydopamine rat model of Parkinsonism. J Neurosci Res. 2005;81(6):865-73. [CrossRef]
- Sonsalla PK, Coleman C, Wong LY, Harris SL, Richardson JR, Gadad BS, et al. The angiotensin converting enzyme inhibitor captopril protects nigrostriatal dopamine neurons in animal models of parkinsonism. Exp Neurol. 2013;250:376-83. [CrossRef]
- Fillit HM, Rockwood K, Young JB (eds). Brocklehurst’s textbook of geriatric medicine and gerontology. 8th ed. Philadelfia: Saunders Elsevier; 2016.
- Abbott A. Neuroscience: The plaque plan. Nature. 2008;456:161-4. [CrossRef]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebocontrolled phase I trial. Lancet. 2008;372:216-23. [CrossRef]
- Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537:50-6. [CrossRef]
- Waite LM. Treatment for Alzheimer’s disease: has anything changed? Aust Prescr. 2015;38(2):60-3. [CrossRef]
- Mendiola-Precoma J, Berumen LC, Padilla K, Garcia-Alcocer G. Therapies for Prevention and Treatment of Alzheimer’s Disease. Biomed Res Int. 2016;2016:2589276; doi:10.1155/2016/2589276. [CrossRef]
- Ballard C, Hanney ML, Theodoulou M, Douglas S, McShane R, Kossakowski K, et al. (2009) The dementia antipsychotic withdrawal trial (DART-AD): long-term follow-up of a randomised placebo-controlled trial. Lancet Neurol. 2009;8:151-7. [CrossRef]
- Fossel MB. The telomerase revolution. Dallas: BenBella Books; 2015.
- Harley CB, Liu W, Blasco M, Vera E, Andrews WH, Briggs LA, Raffaele JM. A natural product telomerase activator as part of a health maintenance program. Rejuvenation Res. 2011;14(1):45-56. [CrossRef]
- Harley CB, Liu W, Flom PL, Raffaele JM. A natural product telomerase activator as part of a health maintenance program: metabolic and cardiovascular response. Rejuvenation Res. 2013;16(5):386-95. [CrossRef]
- Harrison MM, Jenkins BV, O’Connor-Giles KM, Wildonger J. A CRISPR view of development. Genes Dev. 2014;28(17):1859-72. [CrossRef]
- Zhang F, Wen Y, Guo X. CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum Mol Genet. 2014;23(R1):R40-6. [CrossRef]
- Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490-5. [CrossRef]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84-8. [CrossRef]
- O’Geen H, Yu AS, Segal DJ. How specific is CRISPR/Cas9 really? Curr Opin Chem Biol. 2015;29:72-8. [CrossRef]
- Chandrasegaran S, Carroll D. Origins of programmable nucleases for genome engineering. J Mol Biol. 2016;428(5 Pt B):963-89. [CrossRef]
- De Grey A. The SENS challenge: $20,000 says the foreseeable defeat of aging is not laughable. Rejuvenation Res. 2005;8(4):207-10. [CrossRef]
- Skulachev VP. Aging is a specific biological function rather than the result of a disorder in complex living systems: biochemical evidence in support of Weismann's hypothesis. Biochem (Mosc). 1997;62:1191-5.
