

Neuroimaging in Dementia
Kevin T Ong 1, 2, 3, * ![]()
- Armadale Health Service, Mount Nasura, Australia
- Dementia Support Australia, Wembley, Australia
- University of Melbourne, Melbourne, Australia
* Correspondence: : Kevin T Ong ![]()
Academic Editor: Michael Fossel
Received: May 15, 2018 | Accepted: August 28, 2018 | Published: September 04, 2018
OBM Geriatrics 2018, Volume 2, Issue 3 doi:10.21926/obm.geriatr.1803011
Recommended citation: Ong KT. Neuroimaging in Dementia. OBM Geriatrics 2018;2(3):011; doi:10.21926/obm.geriatr.1803011.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Amyloid positron emission tomography (PET), fluorodeoxyglucose (FDG) PET, and magnetic resonance imaging (MRI) are three modalities in the imaging of Alzheimer’s disease (AD). This article reviews the experience and data obtained from AD research using these techniques to understand the natural history of AD and to enhance therapeutic trials. It is now realised that their use can improve early detection of AD, and has opened a new corridor which may lead toward successful treatment.
Keywords
Neuroimaging; dementia; Alzheimer's disease; amyloid imaging; positron emission tomography; MRI
1. Introduction
Prior to the last decade, Alzheimer’s disease (AD) neuropathology was only visible and measurable in vitro using brain tissue from autopsy or, less commonly, from brain biopsy. Developing biomarkers that distinguish AD from non-AD dementia was and still is a major need [1]. Positron emission tomography (PET) tracers that bind directly to proteinopathies in vivo enable their detection even when accumulation is in the early phase. It became possible in the last decade to reliably detect Aβ amyloid plaques in vivo with PET, long before dementia due to AD develops, and with high correlation to histopathological measures of amyloid. This now provides a long window of opportunity to potentially intervene and alter the progression of AD before major neuronal damage has occurred. With the surge in dementia research using amyloid PET, FDG PET and magnetic resonance imaging (MRI) over the last decade, there is now sufficient experience in the imaging of AD with these three modalities, in both its natural history and therapeutic trials, to have confidence that their use opens a new corridor which may lead toward successful treatment [2,3].
Neuroimaging is able to measure several events in the amyloid cascade. For example, increased Aβ42 production and/or decreased elimination results in the deposition of accumulated soluble Aβ42 oligomers as fibrillar plaques, which can be detected by β-amyloid PET imaging. Aβ42 oligomers exert subtle effects on synapses resulting in progressive synaptic and neuritic injury, altered neuronal ionic homeostasis and oxidative injury, altered kinase and phosphatase activities and tau tangle formation, of which the latter can be detected by tau PET. This results in widespread neuronal dysfunction which is detected by hypometabolism on FDG PET, and cell death which can be detected by atrophy on MRI.
The escalating number of studies involving Aβ imaging, and the relatively fewer studies on neurofibrillary tangle (NFT) imaging due to its later tracer development, raises the concern that Aβ is overemphasized as the key driver of cognitive decline in AD. However, the stronger proximal association between tauopathy and neurodegeneration in contrast to the long time lag between Aβ positivity and neurodegeneration [4], might render tau PET to be not as promising as Aβ PET in improving clinical detection in the pre-dementia stages.
2. Cerebral Aβ Amyloid Imaging with Positron Emission Tomography
2.1 Introduction
The team at the university of Pittsburgh was the first to develop a tracer used in amyloid imaging. Pittsburgh University Professors William Klunk, a psychogeriatrician, and Chester Mathis, a radiochemist, were originally looking for a faster way to find amyloid fighting drugs, so they devised a tracer that could image amyloid. After a journey of more than 10 years, they published their initial paper in 2004 on their ground breaking 11C-Pittsburgh compound B (11C-PiB) tracer [5].
The concept of amyloid imaging is straightforward. Dyes that bind to plaques in post mortem brain tissue were modulated to be able to cross the blood brain barrier, and tagged with a positron emitting radioactive molecule. The radioactive tracer is injected intravenously at a micromolar concentration and the patient is placed in a PET scanner. Using coincidence detection of the paired gamma rays emitted by positron annihilation, the PET scanner creates a three dimensional image of brain radioactivity that reflects where and how much tracer has stuck to amyloid plaque.
Amyloid detected on 11C-PiB PET scan is associated with fibrillar amyloid pathology. 11C-PiB does not pick up cortical Lewy bodies [5]. Increased amyloid 11C-PiB PET tracer uptake may also occur in a few non-AD clinical syndromes where beta-amyloid aggregates are present like dementia with Lewy bodies [6], and cerebral amyloid angiopathy [7]. Familial AD usually have very high uptake in the basal ganglia area compared to the distribution seen in sporadic AD [8].
The United States Federal Drug Agency has approved three 18F-labelled β-amyloid PET tracers for clinical use to rule out AD in suspected AD cases. Florbetapir was approved in 2012. Flutemetamol was approved in 2013. Florbetaben was approved in 2014. A negative scan using these traces indicates sparse to no neuritic plaques and makes the diagnosis of AD unlikely. A positive scan indicates moderate to frequent amyloid neuritic plaques and while supporting a diagnosis of AD, it does not necessarily mean that the presenting symptoms are due to AD. It is possible that the patient could still be in the long period of asymptomatic amyloid accumulation and current symptoms are due to another disorder. Figure 1.

Figure 1 [9] Sagittal and axial 18F-Florbetaben PET images of two mild cognitive impairment research participants (figure taken from Ong, 2013). Both were 73 years old and had the same Mini-Mental State Examination score of 27 out of 30. The PET images on the left show non-specific retention in white matter. The PET images on the right show high cortical tracer retention in the typical pattern seen in Alzheimer’s disease. The participant with PET images on the right progressed to Alzheimer’s disease within two years while the participant with PET images on the left did not.
2.2 Comparison Studies
Amyloid imaging has been shown in clinical Phase III studies to correlate well with Aβ neuropathology. Published reports from these Phase III studies comparing binary visual readings of images taken during life to autopsy findings in those that died less than 2 years later, for confirming significant plaque load (i.e. CERAD criteria of moderate or frequent neuritic plaques), showed florbetapir (n=59) had a 97% sensitivity and a 100% specificity [10,11], flutemetamol (n=68) had a 88% sensitivity and 88% specificity [12], and florbetaben (n=74) had a 98% sensitivity and a 89% specificity [13]. Altogether there is over 90% sensitivity and specificity on average for the F-18 labelled amyloid tracers in subjects who had histopathology (n=201).
In order to compare studies using different amyloid imaging tracers, head-to-head comparisons of different tracers in the same subjects should be carried out. To date, there have only been head-to-head comparisons published of florbetaben and of a non-FDA approved 18F labelled tracer, NAV4694 with 11C-PiB [14,15,16,17]. Attempts have been made to use available data to make meaningful comparisons [18]. Differences in the non-specific white matter uptake and the tracer affinity for cortical plaques between the tracers are apparent but the influence of this on detection and monitoring of beta-amyloid plaques remains unclear without head-to-head studies.
2.3 Practical Applications and Caveats
Amyloid PET use has been shown to impact clinical decision making. In one study of 229 individuals with cognitive decline and uncertain diagnosis, physicians made changes in diagnoses in 56% after their PET scan [19]. Diagnostic confidence increased by an average of 21.6%. 86.9% of cases had at least one change in their management plan. Acetylcholinesterase Inhibitor or Memantine use increased by 17.7% among amyloid positive cases, and decreased by 23.3% among those with negative scans. Planned structural brain imaging decreased by 24.4%, and planned neuropsychological testing decreased by 32.6%. However the diagnostic yield and prognostic utility of amyloid PET decreases with age due to increasing prevalence of amyloid positivity and competing co-morbidities with advancing age [20,21].
Methods used in assessing an amyloid PET scan are varied [7]. Approaches include automated reading, qualitative visual reading based on five cortical regions and ventral striatum, semi-quantitative regional standardised uptake value ratio (SUVR) analysis, semi-quantitative global SUVR analyses, and Logan distribution volume ratio (DVR). Different approaches may result in different results in those scans which have an intermediate level of tracer uptake. Time activity curve studies show that the tracers have nearly identical uptake and fast clearance from the cerebellar cortex in both healthy controls and mild or moderate dementia severity AD patients, consistent with the absence of neuritic Aβ plaques in this region before late AD. While use of Logan DVR gives better precision of signal strength and lower noise than SUVR, DVR is far more time consuming to obtain as it requires a continuous scan of at least 60 minutes [22]. Notwithstanding, SUVR measured from a twenty minute scan commenced 90 minutes post injection of tracer for florbetaben has excellent correlation with DVR, and is precise enough for research use [23].
Aβ burden is a continuous process, and should not be thought of as dichotomous. Nonetheless, studies often divide subjects into those with high and low Aβ burden for comparisons. In order to better standardise and compare results in prospective trials, it is useful to have an established reference value to serve as a “cutoff”. The practical difficulties with “cutoffs” mainly revolved around those subjects whose scan results are very close to the “cutoff” value, and so end up with a somewhat ambiguous result. Should such an event occur, then individual regions of interest should be reviewed to see if any AD vulnerable regions (frontal, temporal, parietal, posterior cingulate-precuneus) have high uptake. If so, then the scan should be considered positive.
Several amyloid imaging studies have shown test/re-test reproducibility of up to 95%. A six percent or more change in SUVR in the same individual on serial florbetaben scans could be considered genuine if considered as a single case alone [23].
One study compared amyloid imaging results in 32 individuals from the Alzheimer’s disease neuroimaging initiative cohort with both florbetapir and 11C-PiB scans acquired on a variety of scanner types, and processed using a variety of methods [15]. They found that the scale of cortical retention ratios was not affected by image preprocessing or analysis methods, but they were affected predominantly by the type of radioligand and the reference region used for scanning. Both ligands had excellent agreement in categorizing the patients as either amyloid positive or amyloid negative.
In longitudinal amyloid imaging, not correcting for effects of brain atrophy with aging or disease progression will lower measures such as SUVR and DVR due to partial volume effect, a consequence of the resolution limitations of PET imaging. MRI measurement of cortical volume can be used to correct partial volume effect though this may introduce some “noise” into the amyloid measurement. Some patients with AD do accumulate cerebellar Aβ plaques in late AD, and this may result in a further underestimation of cortical plaque load using cortex to cerebellar ratio measures such as SUVR and DVR. Theoretically it is possible that suitable surfaces for oligomers to incorporate into existing plaques might be exhausted in late AD. Cerebral spinal fluid (CSF) Aβ have been shown to increase over time in AD with low levels [24], possibly signalling that it is not assimilating into Aβ plaques. These features may explain why Aβ accumulation appears to slow or even decrease on serial amyloid PET scans in the later stages of AD.
2.4 Guidelines for Clinical Use
To provide guidance to dementia care practitioners, patients, and caregivers, the Alzheimer’s Association and the Society of Nuclear Medicine and Molecular Imaging convened the Amyloid Imaging Taskforce (AIT) [7]. The AIT considered a broad range of specific clinical scenarios in which amyloid PET could potentially be used appropriately.
The AIT felt that amyloid imaging is appropriate if after completing a full cognitive work-up and evaluation by a dementia expert, (1) there is a cognitive complaint with objectively confirmed impairment, and (2) AD is a possible diagnosis but the diagnosis is uncertain, and (3) when knowledge of the presence or absence of Aβ pathology is expected to increase diagnostic certainty and alter management, and (4) if ordered by dementia experts involved in dementia care provision due to complexities in the field. Hence amyloid imaging is appropriate in approved settings, when patients present with persistent or progressive unexplained mild cognitive impairment (MCI), when patients satisfy core clinical criteria for only possible and not probable AD because of unclear clinical presentation due to either an atypical clinical course or an etiologically mixed presentation, and when patients present with progressive dementia and atypically early age of onset, usually defined as 65 years or less in age. A simple and efficient way of considering whether an amyloid scan is appropriate is to apply Bayesian logic, by considering whether the post test probability of AD could be significantly changed by the scan result, in the context of the patient’s situation, and whether this will change management or help the treating clinician to be more confident in treatment choices.
Amyloid imaging is considered inappropriate by the AIT in the following situations:
1. Patients with core clinical criteria for probable AD with typical age of onset.
2. To determine dementia severity.
3. Based solely on a positive family history of dementia or presence of Apolipoprotein E ε4.
4. Patients with a cognitive complaint that is unconfirmed on clinical examination.
5. In lieu of genotyping for suspected autosomal dominant mutation carriers.
6. In asymptomatic individuals.
7. Non-medical use (e.g., legal, insurance coverage, or employment screening).
Amyloid imaging is now available for clinical use, and indications and guidelines have been defined. Should successful disease modification become a widely available reality, PET amyloid imaging is likely to move very rapidly into routine clinical practice. At present, the high cost of amyloid PET imaging, the lack of reimbursement by governments and insurance companies, and the limited scope for altering disease outcome due to the absence of disease modifying therapy, see amyloid PET rarely used in clinical practice.
The clinical benefits of better detection of AD pathology and earlier, more accurate diagnosis are limited in the absence of disease modifying therapy. However, in addition to providing diagnostic clarity that may reduce carer and patient stress and allow better lifestyle decisions and appropriate preparation for the future, amyloid PET may guide the appropriate use of medications such as the Acetylcholinesterase Inhibitors that can significantly exacerbate symptoms if given to patients with frontotemporal dementia (FTD) [25].
3. Structural Brain Imaging with Magnetic Resonance Imaging
3.1 Introduction
Structural imaging began with computed tomography (CT) but has been surpassed by MRI due to the better tissue contrast of the latter allowing more accurate assessment of grey matter volume and more sensitive detection of vascular and other pathological changes. MRI is used routinely in dementia studies for subject recruitment to exclude confounding medical conditions, for determining whether there is an effect of treatment on disease progression, and to detect vasogenic oedema in immune-based treatments as a safety end point. Screening for reduced hippocampal volume with MRI enables a more homogeneous cohort of rapidly declining subjects, reducing variability in longitudinal outcome measures.
MRI is sensitive for detection of vascular disease that may contribute to cognitive impairment. The primary issue with volumetric MRI as a diagnostic measure for AD or outcome measure in clinical trials is that change in volume is non-specific. Volume loss in neurodegeneration can be related to NFT or other processes. It is unclear whether or how much the increased rate of cortical atrophy and ventricular enlargement seen in longitudinal studies of AD and clinical trials reflects increased neurodegeneration, amyloid removal, reduction in amyloid associated inflammation, or changes in CSF absorption. Although hippocampal volume correlates with neuron counts [26,27]. and Braak stage in AD [28], hippocampal atrophy can be associated with different types of neuropathology like cerebrovascular disease, FTD, and hippocampal sclerosis, and so is not specific for AD [28,29].
Despite the limitations of MRI biomarkers, structural imaging with MRI is still necessary to rule out specific causes of cognitive decline, like stroke, tumour, or hydrocephalus. While cerebral atrophy detected on MRI is non-specific, it is more closely related to cognition than amyloid PET, since the neurodegeneration curve is closer to cognitive decline compared to amyloid load. Therefore MRI is better for evaluating disease progression, not predicting it. Also by broadly indicating the regions of atrophy and degree of atrophy in individual regions, a clinical MRI can offer some sense of what the pathophysiology of the cognitive impairment is. So in non-early cases with a typical history for AD, simpler measures like MRI alone may be sufficient to aid in diagnosing AD by confirming an AD pattern of atrophy and excluding other structural causes which are infrequent.
3.2 Leveraging Routine Magnetic Resonance Imaging in the Clinical Assessment of Dementia
It is possible to leverage structural MRI performed in the routine clinical assessment of patients by qualitatively looking for pathognomic atrophy patterns for AD, in particular the combination of relatively more pronounced atrophy in the posterior cingulate-precuneus region and bilateral atrophy in the hippocampus. The posterior cingulate-precuneus is the first area to be affected by amyloid deposition [30], and the hippocampus is the first area to be affected by neurofibrillary tangles in AD. The acceleration of atrophy before the first clinical sign is especially marked in these two regions [31]. Hence this atrophy combination pattern is closely associated with poor memory due to AD. Note that the hippocampal atrophy should extend to the temporal neocortex, and is not predominant to either side, to be more specific for AD. Otherwise alternate pathology has to be considered. Finally, frontal variant AD tends not to have predominantly frontal atrophy unlike behavioural variant FTD [32]. Figure 2.

Figure 2 [32] Brain Atrophy Patterns (figure taken from Ossenkoppele, 2015). Frequency maps of the proportion of patients within each diagnostic group with subthreshold values compared to a group of healthy controls. Hot colours indicate involvement of various brain regions. The posterior cingulate region is particularly affected in Alzheimer’s disease regardless of phenotype.
On top of the pathognomic AD atrophy pattern, the presence of three or more microhaemorrhages in the posterior lobar deep grey (rather than infratentorial) regions may indicate amyloid angiopathy, a finding that is common in patients with AD [33]. Cerebral microhaemorrhages are seen as small, dark areas, on gradient echo or susceptibility weighted imaging MRI, which can be routinely and quickly obtained. They can at times also be seen on CT. An elderly person with amnestic concerns who has more than three microhaemorrhages in the posterior cortices without significant infarct, lacunes, microhaemorrhages in other regions or white matter disease is likely to have cerebral amyloid angiopathy due to AD pathology [34,35,36]. Microhaemorrhages in vascular disease tend to be seen in the deep or infratentorial location like the basal ganglia where arteries are more affected by hypertension. While amyloid angiopathy occurs with age, in the setting of cognitive impairment, microhaemorrhages tend to be associated with amyloid pathology particularly in Apolipoprotein E ε4 carriers, men more than women and hypertension. Figure 3.
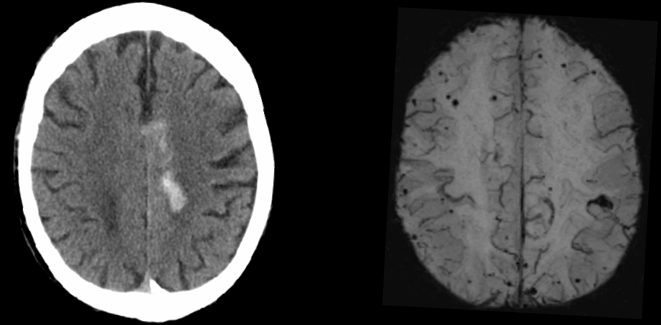
Figure 3 Left: Brain CT axial image of an elderly patient presenting with a haemorrhagic stroke on a background of progressive cognitive decline. Right: Brain (SWI) MRI axial image of the same patient done a few days later showing extensive microhaemorrhages.
3.3 Medial Temporal Atrophy
The most studied structure in AD with MRI is the hippocampus [37,38,39,40,41,42]. Hippocampal atrophy has over 70% accuracy in classifying AD patients from control groups, and MCI who would convert to AD within 12-18 months compared to normal [37,38]. Following initial descriptions of hippocampal atrophy using MRI [43,44], a further series of studies showed that rate of atrophy increased by clinical groups from normal control to MCI to AD, consistent with an exponential decrease in neuronal and synaptic loss that parallels cognitive decline. The annual rate of hippocampal atrophy in MCI is approximately 3.5%, and in MCI who convert to AD, the rate increases by approximately 0.22% [45]. Hippocampal atrophic rates are 1 to 1.5% higher for women than men [46]. Although hippocampal atrophy is considered a biomarker of AD, several studies have consistently suggested limited added utility for MRI hippocampal volumes to clinical assessment of memory and function in predicting transition from MCI to AD [47]. Automated hippocampal segmentation has been the focus of development, yet manual segmentation remains the gold standard [48]. Scales for visual assessment of medial temporal atrophy (MTA) have been quantified [49,50]. MTA visual reads work reasonably well under the age 75, but are not specific for AD in patients over 75. There is a strong correlation between visual assessment of MTA by radiologists and quantified hippocampal volume, and expert radiological assessment has been reported to be as accurate as computer-based volumetry for classification of AD [51].
Voxel based MRI studies have demonstrated that atrophy spreads from the medial temporal lobe to the parietal, occipital, and frontal lobes over the course of AD, with MCI who convert to AD, having a more AD-like pattern of atrophy at baseline [48]. The characteristic AD atrophy pattern appears to plateau in MCI non-converters, but progresses in MCI converters and AD patients [52]. Less impaired MCI patients have the greatest rates of atrophy in the medial temporal cortex, where-as more impaired MCI have the greatest rates of atrophy in the prefrontal, parietal, and anterior regions [52,53,54,55,56,57,58].
The volumes of the entorhinal cortex, lateral ventricles, and hippocampus have the greatest effect sizes for distinguishing young healthy controls and AD [52], making them potential biomarkers for the early detection of AD. In MCI who convert and in AD subjects, highest rates of atrophy occur in the hippocampus and entorhinal cortex [46,57,59]. MRI studies have shown progressive cortical atrophy from normal control subjects to single domain MCI to multi domain MCI and finally AD subjects in cross-sectional studies [40,60]. One study showed that the hippocampus and entorhinal cortex have the largest effect size between the control group and the single domain MCI group. Multi domain MCIs had greater differences, as well as differences in the lateral inferior, middle, and superior temporal gyri and fusiform cortices relative to controls.
MRI studies show that global atrophy is faster in those with cortical amyloid [61]. Posterior atrophy is associated with a positive amyloid PET scan, where as anterior atrophy is associated with a negative amyloid PET scan [62]. In subjects who develop MCI, their MRI trajectories change faster than those who remain normal [63]. Also there is an acceleration of trajectory close to cognitive decline, and this is measurable. The rate of hippocampal atrophy on MRI is greater than two times in AD compared to controls, and higher in converters to MCI or AD compared to stable or non-converters [64]. Cross-sectional and longitudinal studies consistently report a strong relationship between rate of atrophy and rate of cognitive decline with anatomical and functional specificity. In MCI converters the parietal and temporal areas have most atrophy compared to non-converters [65], and conversion to AD is associated with disease effects on the default mode network and the prefrontal and medial temporal structures [66].
Currently there are no standardized MRI imaging parameters and methods of volumetric assessment. These are required to develop specific cutoffs for hippocampal atrophy, so as to improve comparisons between studies and predictive accuracy. While brain atrophy has discriminatory power, multiple sophisticated methods of structural analyses of MRI give varying degrees of accuracy [48,64,67]. The choice of protocol for automatic multitemplate-based segmentation can influence segmentation accuracy [68]. Hence comparing results from different studies that use different methodologies is problematic.
3.4 White Matter Hyperintensities
On top of atrophy, infarcts and microhaemorrhages, MRI can also detect white matter hyperintensities (WMH). While hippocampal volume decreases with age, abnormal periventricular white matter signals increase with age, worse with more vascular risk factors. Over 95% of individuals above the age of 65 have some white matter disease and this is usually associated with vascular risk factors [69,70]. A sub-study of the Perindopril Protection against Recurrent Stroke Study revealed less progression of WMH with aggressive antihypertensive treatment [71]. A threshold of 10cc of periventricular WMH is needed to see cognitive effects [72]. Those with extensive white matter disease have poorer global cognitive function, executive function, working memory, processing speed, gait, psychomotor speed, and dexterity [73,74,75]. Extensive WMH is associated with MCI and an increased risk of stroke, dementia, and death [76,77]. Twin studies suggest a genetic contribution to WMH [77].
The cause of WMH is likely to be multifactorial. It has been postulated that WMH can be accounted for by: (1) arterioles becoming tortuous with aging [78], causing stress due to underperfusion, which leads to collagenous formation, and WMH on MRI, (2) leakage of fluid around diseased vessels and loss of blood brain barrier integrity, and (3) severe neuronal drop out as frequently seen on MRI in frontotemporal dementia and autosomal dominant AD.
Baseline WMH volume does not predict changes in CSF biomarkers, glucose metabolism, or hippocampal atrophy as it is not specific to AD [79]. Higher WMH volume is associated with cerebrovascular disease and contributes to cognitive impairment independent of other neuropathology [80].
4. Brain Fluorodeoxyglucose Positron Emission Tomography
4.1 Introduction
Co-morbid pathology is common in autopsy studies. For example, in one consecutive series of autopsies, only four out of 22 had pure AD [81]. 10 had coincident dementia with Lewy bodies (DLB), nine had mesial temporal pathology (TDP-43 proteinopathy, argyrophilic grain disease, and hippocampal sclerosis), and five had vascular pathology. Nonetheless, biomarkers like FDG PET enables us to look for typical patterns of hypometabolism that are associated with neurodegenerative disease subtypes. For example, by showing occipital hypometabolism, FDG PET can predict coincident DLB in AD patients [48]. Figure 4.
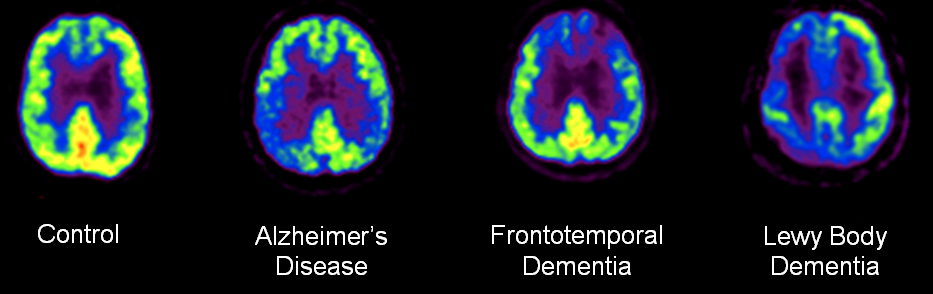
Figure 4 18F-Fluorodeoxyglucose PET detecting common neurodegenerative diseases. These axial images were obtained from the Austin Hospital Nuclear Medicine Department, Australia.
FDG PET measures glucose metabolism. Glucose hypometabolism reflects neural communication between neurons and therefore the degree of neuron dysfunction [82,83]. FDG hypometabolism correlates with NFT [84]. Both MCI and AD patients have lower metabolism in bilateral posterior cingulate-precuneus and parietotemporal regions [85], and the magnitude and spatial extent is greater with increased disease severity [86]. Reductions in glucose metabolism occur in the posterior cingulate, then medial and lateral parietal and medial and lateral temporal cortex [87], but increasingly involves frontal cortex after the onset of dementia [86]. Hence impaired memory in MCI is associated with reduced metabolism in the parietal and temporal lobes, where as severely impaired memory in AD may better correlate with the degree of frontal and orbital frontal hypometabolism [88].
4.2 Clinical Impact
Studies have shown that in the clinical setting, FDG PET results have led to clinicians to change their clinical diagnoses a quarter of the time when diagnostic certainty was under 90% pre-PET [89]. Diagnostic confidence in this study also increased from 71% pre-PET to 87% post PET. A prospective study of 194 consecutive memory clinic patients who were referred for a FDG PET at the discretion of the dementia specialist showed that management impact was higher in patients with greater diagnostic uncertainty and in those with less severe cognitive impairment [90].
Although there is a correlation between hypometabolism and cognitive measures of disease severity, hippocampal volume and CSF biomarkers [91], the relationship between glucose hypometabolism and cognitive decline is largely independent from other biomarkers [92]. Hence the addition of FDG PET to the clinical work-up could provide complimentary information to improve assessment of disease proximity.
In studies that have used prolonged clinical follow-up or autopsy diagnosis as the standard of truth, FDG PET had an accuracy of 93% distinguishing between AD and healthy controls, and 85% distinguishing between AD and other dementias [93]. Clinical visual reads have been shown to increase in accuracy with the use of surface based displays of voxel Z-scores derived from comparison to a normal database such as used in the three dimensional stereotactic surface projection approach.
4.3 Caveats
FDG PET can aid diagnosis but must be interpreted in clinical context. Hypometabolism can develop before the appearance of cognitive decline [94]. In using FDG PET to assist with clinical assessment, we need to consider brain areas with hypometabolism and brain areas that are preserved. Hypometabolism in the temporoparietal and posterior cingulate-precuneus regions is quite specific to underlying AD neuropathology. Frontal hypometabolism in addition to these areas discriminates MCI converters from non-converters. General hypometabolism is not specific to AD or a neurodegenerative subtype. The degree of hypometabolism is greater in MCI converters compared to non-converters [48,95].
FDG metabolism has been reported to increase in healthy elderly and patients with early MCI who have positive amyloid PET scans [96,97]. This may reflect a ‘compensatory’ hypermetabolic phase, inflammation, or it may be hyper-excitation due to neuronal toxicity. Bidirectional FDG changes in early disease may complicate the detection of metabolic abnormalities.
5. Amalgamating Multimodal Biomarkers
5.1 Introduction
No single imaging technique will serve all purposes, and each technique should be used in conjunction with other techniques to help researchers gain insight into the pathogenesis of AD. Also, the availability of biomarkers should not restrict sound clinical judgement. It is important to regard Aβ amyloid-imaging as a technique that is complementary to structural (e.g. MRI, CT), functional (e.g. FDG PET, functional MRI) and other molecular (e.g. DAT, tau) imaging techniques. Considerable work needs to be conducted to establish what tests and profiles are better for which particular time points in the disease. Ultimately the best diagnostic approach involves a full clinical work-up, that must consist of a good clinical and collateral history, sensitive and specific cognitive tests, MRI, and where required, biological markers such as Aβ imaging or CSF Aβ42 and tau. Figure 5 and Table 1.
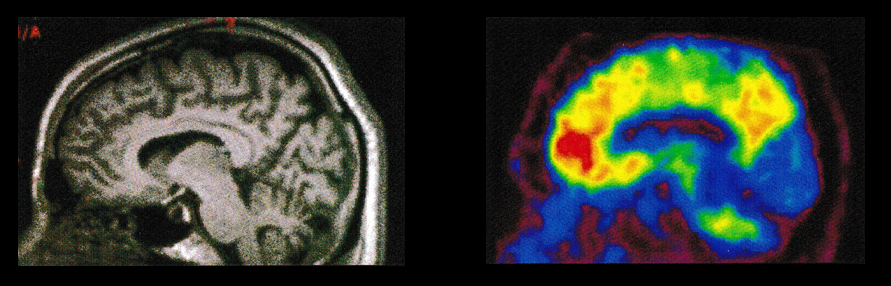
Figure 5 Left: Brain MRI sagittal image in a patient with possible Alzheimer’s disease. Right: Brain amyloid PET sagittal image confirms the cerebral β-amyloid build up in this patient’s brain.
5.2 Disease Stages and Accuracy
Biomarkers have varying degrees of steepness of rates of change in different disease stages. This causes an inconsistent association between Aβ and cross-sectional neurodegeneration [98,99,100,101,102,103,104,105,106,107]. For example, CSF Aβ42 levels decline most rapidly in preclinical (asymptomatic) AD, where as glucose metabolism declines most rapidly in the dementia phase of AD [42]. When individuals progress into the dementia phase, both the FDG metabolism and hippocampal volume curves drop rapidly. In general, there is local correlation in the posterior cingulate-precuneus and temporoparietal regions between hypometabolism and atrophy [108,109,110]. However, the degree of hypometabolism exceeds the degree of atrophy in the posterior cingulate-precuneus and medial orbital frontal regions and persists after partial volume correction. By contrast the degree of hippocampal atrophy predominates or is similar to the degree of hypometabolism. Due to the discrepancies between Aβ and cross-sectional neurodegeneration, multimodal biomarkers increase prognostic power and diagnostic accuracy [111]. Finally, CSF Aβ42 and amyloid PET have been hypothesized to measure different aspects of Aβ pathology in AD with CSF revealing the more soluble components and Aβ imaging with amyloid PET predominantly reflecting dense plaque [112].
Table 1 Summary table of brain MRI, FDG PET and amyloid PET use in early dementia work-up
|
|
Characteristics |
Advantages |
Disadvantages |
|
MRI |
|
|
|
|
FDG PET |
|
|
|
|
β-amyloid PET |
|
|
|
Amyloid markers signal disease initiation and early progression with rate of accumulation declining in late stages of the illness. FDG PET measures of glucose hypometabolism and CSF tau levels increase later but still relatively early in disease progression and throughout the stages of AD. Hippocampal atrophy in AD increases later in disease progression [113]. Hence Aβ measures associated with early disease have greater changes in non-demented patients where as FDG PET changes are more associated in AD [114]. One longitudinal study found that Aβ can have an effect on brain structure and function independent of CSF tau, and CSF tau can have an effect on baseline cognition independent of neuroimaging measures [115].
Amyloid imaging is specific for Aβ neuropathology. Amyloid scans tend to be easier to read than FDG scans. One study found that in distinguishing between AD (n=62) and FTD (n=45) by visual reads, 11C-PiB had a sensitivity of 90% and a specificity of 83% where as FDG PET had a sensitivity of 78% and a specificity of 84%. 11C-PiB had better inter-rater agreement (k=0.96 vs k=0.72), and was more sensitive in detecting AD pathology (n=12, 11C-PiB 97%; n=10, FDG 87%) [116]. However, as fluorine substitutions greatly increase a molecule’s lipophilicity, the FDA approved F-18 labelled amyloid tracers have higher non-specific sub-cortical white matter (i.e. more lipid rich area) binding compared to 11C-PiB.
5.3 Practical Challenges
Considerable challenges remain in establishing the relationship between biological and cognitive measures throughout the chronology of the preclinical phase of AD. As the field progresses towards a deeper understanding of the evolution of AD neuropathology in the pre-dementia stage, through the improved ability to more precisely detect longitudinal changes in the presence and amount of Aβ accumulation, glucose hypometabolism, and atrophy in vivo, the prospect of implementing and monitoring disease-modifying treatments for AD to prevent further evolution has become more realistic. Amyloid PET can be used both to improve patient recruitment and as a secondary end point measure in disease modifying drug trials. By comparing to a control group, it can be shown whether a drug is successful in stabilising or reducing Aβ accumulation over time, and whether this is associated with preservation or improvement in cognition. Brain FDG PET and MRI are non-specific measures of neurodegeneration, therefore they are not considered good in vivo markers of specific target engagement for amyloid based disease modifying treatment.
6. Final Word
To conclude, we now have the means to enable early and accurate diagnosis in dementia. While we may not be able to eliminate the impact of the coming dementia epidemic, early detection offers us the opportunities to reduce it significantly. Even so another major challenge facing health care providers is stability of funding for patient access to care, and training health care workers in incorporating new developments to improving early diagnosis. Providing scans to all those at risk in the community involves significant infrastructure and cost considerations. Nonetheless, should disease modifying therapy become available, the diagnostic potential of dementia neuroimaging will be fully realised, and the issue of expanding infrastructure to meet the demands will be a subject of further debate.
Author Contributions
This paper was originally written as a chapter in the author’s doctorate thesis ‘Tracking Cerebral Amyloid Accumulation In Ageing, Mild Cognitive Impairment, Alzheimer’s Disease, and Other Dementias’, and the author is the sole author on this paper.
Competing Interests
The author has declared that no competing interests exist.
References
- Shaw LM, Korecka M, Clark CM, Lee VM and Trojanowski JQ. Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat Rev Drug Discov. 2007; 6: 295-303. [CrossRef] [Google scholar] [PubMed]
- Hill D. Neuroimaging to assess safety and efficacy of AD therapies. Expert Opin Investig Drugs. 2010; 19: 23-26. [CrossRef] [Google scholar] [PubMed]
- Weiner MW. Imaging and biomarkers will be used for detection and monitoring progression of early Alzheimer's disease. J Nutr Health Aging. 2009; 13: 332. [CrossRef] [Google scholar] [PubMed]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012; 71: 362-381. [CrossRef] [Google scholar] [PubMed]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004; 55: 306-319. [CrossRef] [Google scholar] [PubMed]
- Villemagne VL, Ong K, Mulligan RS, Holl G, Pejoska S, Jones G, et al. Amyloid imaging with (18)F-florbetaben in Alzheimer disease and other dementias. J Nucl Med. 2011; 52: 1210-1217. [CrossRef] [Google scholar] [PubMed]
- Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer's Association. J Nucl Med. 2013; 54: 476-490. [CrossRef] [Google scholar] [PubMed]
- Benzinger TLS, Blazey T, Jack CR, Koeppe RA, Su Y, Xiong C, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc Natl Acad Sci U S A. 2013; 110: E4502-E4509. [CrossRef] [Google scholar] [PubMed]
- Ong K, Villemagne VL, Bahar-Fuchs A, Lamb F, Chételat G, Raniga P, et al. 18F-florbetaben Aβ imaging in mild cognitive impairment. Alzheimer's Research & Therapy. 2013; 5: 1-11. [CrossRef] [Google scholar] [PubMed]
- Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. Jama. 2011; 305: 275-283. [CrossRef] [Google scholar] [PubMed]
- Clark CM, Pontecorvo MJ, Beach TG, Bedell BJ, Coleman RE, Doraiswamy PM, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-beta plaques: a prospective cohort study. Lancet Neurol. 2012; 11: 669-678. [CrossRef] [Google scholar] [PubMed]
- Curtis C, Gamez JE, Singh U, Sadowsky CH, Villena T, Sabbagh MN, et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 2015; 72: 287-294. [CrossRef] [Google scholar] [PubMed]
- Sabri O, Sabbagh MN, Seibyl J, Barthel H, Akatsu H, Ouchi Y, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer's disease: phase 3 study. Alzheimers Dement. 2015; 11: 964-974. [CrossRef] [Google scholar] [PubMed]
- Rowe CC, Pejoska S, Mulligan RS, Jones G, Chan JG, Svensson S, et al. Head-to-head comparison of 11C-PiB and 18F-AZD4694 (NAV4694) for beta-amyloid imaging in aging and dementia. J Nucl Med. 2013; 54: 880-886. [CrossRef] [Google scholar] [PubMed]
- Landau SM, Breault C, Joshi AD, Pontecorvo M, Mathis CA, Jagust WJ, et al. Amyloid-beta imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med. 2013; 54: 70-77. [CrossRef] [Google scholar] [PubMed]
- Villemagne VL, Mulligan RS, Pejoska S, Ong K, Jones G, O'Keefe G, et al. Comparison of 11C-PiB and 18F-florbetaben for Abeta imaging in ageing and Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2012; 39: 983-989. [CrossRef] [Google scholar] [PubMed]
- Rowe CC, Jones G, Dore V, Pejoska S, Margison L, Mulligan RS, et al. Standardized expression of 18F-NAV4694 and 11C-PiB beta-Amyloid PET results with the Centiloid Scale. J Nucl Med. 2016; 57: 1233-1237. [CrossRef] [Google scholar] [PubMed]
- Villemagne VL, Doré V, Yates P, Brown B, Mulligan R, Bourgeat P, et al. En attendant centiloid. 2014. [Google scholar]
- Grundman M, Pontecorvo MJ, Salloway SP, Doraiswamy PM, Fleisher AS, Sadowsky CH, et al. Potential impact of amyloid imaging on diagnosis and intended management in patients with progressive cognitive decline. Alzheimer Dis Assoc Disord. 2013; 27: 4-15. [CrossRef] [Google scholar] [PubMed]
- Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FRJ, et al. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA. 2015; 313: 1924-1938. [CrossRef] [Google scholar] [PubMed]
- Ossenkoppele R, Jansen WJ, Rabinovici GD, Knol DL, van der Flier WM, van Berckel BNM, et al. Prevalence of amyloid pet positivity in dementia syndromes: A meta-analysis. JAMA. 2015; 313: 1939-1950. [CrossRef] [Google scholar] [PubMed]
- van Berckel BN, Ossenkoppele R, Tolboom N, Yaqub M, Foster-Dingley JC, Windhorst AD, et al. Longitudinal amyloid imaging using 11C-PiB: methodologic considerations. J Nucl Med. 2013; 54: 1570-1576. [CrossRef] [Google scholar] [PubMed]
- Sabri O, Seibyl J, Rowe C and Barthel H. Beta-amyloid imaging with florbetaben. Clin Transl Imaging. 2015; 3: 13-26. [CrossRef] [Google scholar] [PubMed]
- Toledo JB, Xie SX, Trojanowski JQ and Shaw LM. Longitudinal change in CSF Tau and Abeta biomarkers for up to 48 months in ADNI. Acta Neuropathol. 2013; 126: 659-670. [CrossRef] [Google scholar] [PubMed]
- Mendez MF, Shapira JS, McMurtray A and Licht E. Preliminary findings: behavioral worsening on donepezil in patients with frontotemporal dementia. Am J Geriatr Psychiat. 2007:15:84-87. [CrossRef] [Google scholar] [PubMed]
- Bobinski M, de Leon MJ, Wegiel J, Desanti S, Convit A, Saint Louis LA, et al. The histological validation of post mortem magnetic resonance imaging-determined hippocampal volume in Alzheimer's disease. Neuroscience. 2000; 95: 721-725. [CrossRef] [Google scholar] [PubMed]
- Zarow C, Vinters HV, Ellis WG, Weiner MW, Mungas D, White L, et al. Correlates of hippocampal neuron number in Alzheimer's disease and ischemic vascular dementia. Ann Neurol. 2005; 57: 896-903. [CrossRef] [Google scholar] [PubMed]
- Jack CR, Jr., Dickson DW, Parisi JE, Xu YC, Cha RH, O'Brien PC, et al. Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology. 2002; 58: 750-757. [CrossRef] [Google scholar] [PubMed]
- Jagust WJ, Landau SM, Shaw LM, Trojanowski JQ, Koeppe RA, Reiman EM, et al. Relationships between biomarkers in aging and dementia. Neurology. 2009; 73: 1193-1199. [CrossRef] [Google scholar] [PubMed]
- Leech R and Sharp DJ. The role of the posterior cingulate cortex in cognition and disease. Brain. 2014; 137: 12-32. [CrossRef] [Google scholar] [PubMed]
- Fox NC, Crum WR, Scahill RI, Stevens JM, Janssen JC and Rossor MN. Imaging of onset and progression of Alzheimer's disease with voxel-compression mapping of serial magnetic resonance images. Lancet. 2001; 358: 201-205. [CrossRef] [Google scholar] [PubMed]
- Ossenkoppele R, Pijnenburg YA, Perry DC, Cohn-Sheehy BI, Scheltens NM, Vogel JW, et al. The behavioural/dysexecutive variant of Alzheimer's disease: clinical, neuroimaging and pathological features. Brain. 2015; 138: 2732-2749. [CrossRef] [Google scholar] [PubMed]
- Chiang GC, Cruz Hernandez JC, Kantarci K, Jack CR, Jr. and Weiner MW. Cerebral microbleeds, CSF p-Tau, and cognitive decline: significance of anatomic distribution. AJNR Am J Neuroradiol. 2015; 36: 1635-1641. [CrossRef] [Google scholar] [PubMed]
- Kalaria RN and Ballard C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord. 1999; 13: S115-S123. [CrossRef] [Google scholar] [PubMed]
- Attems J. Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol. 2005; 110: 345-359. [CrossRef] [Google scholar] [PubMed]
- Cordonnier C and van der Flier WM. Brain microbleeds and Alzheimer’s disease: innocent observation or key player? Brain. 2011; 134: 335-344. [CrossRef] [Google scholar] [PubMed]
- Chupin M, Gerardin E, Cuingnet R, Boutet C, Lemieux L, Lehericy S, et al. Fully automatic hippocampus segmentation and classification in Alzheimer's disease and mild cognitive impairment applied on data from ADNI. Hippocampus. 2009; 19: 579-587. [CrossRef] [Google scholar] [PubMed]
- Wolz R, Heckemann RA, Aljabar P, Hajnal JV, Hammers A, Lotjonen J, et al. Measurement of hippocampal atrophy using 4D graph-cut segmentation: application to ADNI. Neuroimage. 2010; 52: 109-118. [CrossRef] [Google scholar] [PubMed]
- Calvini P, Chincarini A, Gemme G, Penco MA, Squarcia S, Nobili F, et al. Automatic analysis of medial temporal lobe atrophy from structural MRIs for the early assessment of Alzheimer disease. Med Phys. 2009; 36: 3737-3747. [CrossRef] [Google scholar] [PubMed]
- Karow DS, McEvoy LK, Fennema-Notestine C, Hagler DJ, Jr., Jennings RG, Brewer JB, et al. Relative capability of MR imaging and FDG PET to depict changes associated with prodromal and early Alzheimer disease. Radiology. 2010; 256: 932-942. [CrossRef] [Google scholar] [PubMed]
- Lotjonen J, Wolz R, Koikkalainen J, Julkunen V, Thurfjell L, Lundqvist R, et al. Fast and robust extraction of hippocampus from MR images for diagnostics of Alzheimer's disease. Neuroimage. 2011; 56: 185-196. [CrossRef] [Google scholar] [PubMed]
- Lo RY, Hubbard AE, Shaw LM, Trojanowski JQ, Petersen RC, Aisen PS, et al. Longitudinal change of biomarkers in cognitive decline. Arch Neurol. 2011; 68: 1257-1266. [CrossRef] [Google scholar] [PubMed]
- Baron JC, Chetelat G, Desgranges B, Perchey G, Landeau B, de la Sayette V, et al. In vivo mapping of gray matter loss with voxel-based morphometry in mild Alzheimer's disease. Neuroimage. 2001; 14: 298-309. [CrossRef] [Google scholar] [PubMed]
- Seab JP, Jagust WJ, Wong ST, Roos MS, Reed BR and Budinger TF. Quantitative NMR measurements of hippocampal atrophy in Alzheimer's disease. Magn Reson Med. 1988; 8: 200-208. [CrossRef] [Google scholar] [PubMed]
- Leung KK, Bartlett JW, Barnes J, Manning EN, Ourselin S and Fox NC. Cerebral atrophy in mild cognitive impairment and Alzheimer disease: rates and acceleration. Neurology. 2013; 80: 648-654. [CrossRef] [Google scholar] [PubMed]
- Hua X, Hibar DP, Lee S, Toga AW, Jack CR, Jr., Weiner MW, et al. Sex and age differences in atrophic rates: an ADNI study with n=1368 MRI scans. Neurobiol Aging. 2010; 31: 1463-1480. [CrossRef] [Google scholar] [PubMed]
- Devanand DP, Liu X, Brown PJ, Huey ED, Stern Y and Pelton GH. A two-study comparison of clinical and MRI markers of transition from mild cognitive impairment to Alzheimer's disease. Int J Alzheimers Dis. 2012; 483469: 1. [CrossRef] [Google scholar] [PubMed]
- Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Cedarbaum J, et al. 2014 update of the Alzheimer's disease neuroimaging initiative: a review of papers published since its inception. Alzheimers Dement. 2015; 11: e1-e120. [CrossRef] [Google scholar] [PubMed]
- Scheltens P, Launer LJ, Barkhof F, Weinstein HC and van Gool WA. Visual assessment of medial temporal lobe atrophy on magnetic resonance imaging: Interobserver reliability. Journal of Neurology. 1995; 242: 557-560. [CrossRef] [Google scholar] [PubMed]
- Galton C, Gomez-Anson B, Antoun N, Scheltens P, Patterson K, Graves M, et al. Temporal lobe rating scale: application to Alzheimer's disease and frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2001; 70: 165-173. [CrossRef] [Google scholar] [PubMed]
- Boutet C, Chupin M, Colliot O, Sarazin M, Mutlu G, Drier A, et al. Is radiological evaluation as good as computer-based volumetry to assess hippocampal atrophy in Alzheimer's disease? Neuroradiology. 2012; 54: 1321-1330. [CrossRef] [Google scholar] [PubMed]
- Schuff N, Tosun D, Insel PS, Chiang GC, Truran D, Aisen PS, et al. Nonlinear time course of brain volume loss in cognitively normal and impaired elders. Neurobiol Aging. 2012; 33: 845-855. [CrossRef] [Google scholar] [PubMed]
- McDonald CR, McEvoy LK, Gharapetian L, Fennema-Notestine C, Hagler DJ, Jr., Holland D, et al. Regional rates of neocortical atrophy from normal aging to early Alzheimer disease. Neurology. 2009; 73: 457-465. [CrossRef] [Google scholar] [PubMed]
- Hua X, Leow AD, Parikshak N, Lee S, Chiang MC, Toga AW, Jet al. Tensor-based morphometry as a neuroimaging biomarker for Alzheimer's disease: an MRI study of 676 AD, MCI, and normal subjects. Neuroimage. 2008; 43: 458-469. [CrossRef] [Google scholar] [PubMed]
- Leow AD, Yanovsky I, Parikshak N, Hua X, Lee S, Toga AW, et al. Alzheimer's disease neuroimaging initiative: a one-year follow up study using tensor-based morphometry correlating degenerative rates, biomarkers and cognition. Neuroimage. 2009; 45: 645-655. [CrossRef] [Google scholar] [PubMed]
- Risacher SL, Saykin AJ, West JD, Shen L, Firpi HA and McDonald BC. Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort. Curr Alzheimer Res. 2009; 6: 347-361. [CrossRef] [Google scholar] [PubMed]
- Risacher SL, Shen L, West JD, Kim S, McDonald BC, Beckett LA, et al. Longitudinal MRI atrophy biomarkers: relationship to conversion in the ADNI cohort. Neurobiol Aging. 2010; 31: 1401-1418. [CrossRef] [Google scholar] [PubMed]
- McEvoy LK, Fennema-Notestine C, Roddey JC, Hagler DJ, Jr., Holland D, Karow DS, et al. Alzheimer disease: quantitative structural neuroimaging for detection and prediction of clinical and structural changes in mild cognitive impairment. Radiology. 2009; 251: 195-205. [CrossRef] [Google scholar] [PubMed]
- Schuff N, Woerner N, Boreta L, Kornfield T, Shaw LM, Trojanowski JQ, et al. MRI of hippocampal volume loss in early Alzheimer's disease in relation to ApoE genotype and biomarkers. Brain. 2009; 132: 1067-1077. [CrossRef] [Google scholar] [PubMed]
- Fennema-Notestine C, Hagler DJ, Jr., McEvoy LK, Fleisher AS, Wu EH, Karow DS, et al. Structural MRI biomarkers for preclinical and mild Alzheimer's disease. Hum Brain Mapp. 2009; 30: 3238-3253. [CrossRef] [Google scholar] [PubMed]
- Chetelat G, Villemagne VL, Villain N, Jones G, Ellis KA, Ames D, et al. Accelerated cortical atrophy in cognitively normal elderly with high beta-amyloid deposition. Neurology. 2012; 78: 477-484. [CrossRef] [Google scholar] [PubMed]
- Chetelat G, Villemagne VL, Bourgeat P, Pike KE, Jones G, Ames D, et al. Relationship between atrophy and beta-amyloid deposition in Alzheimer disease. Ann Neurol. 2010; 67: 317-324. [Google scholar]
- Carlson NE, Moore MM, Dame A, Howieson D, Silbert LC, Quinn JF, et al. Trajectories of brain loss in aging and the development of cognitive impairment. Neurology. 2008; 70: 828-833. [CrossRef] [Google scholar] [PubMed]
- Jack CR, Jr., Shiung MM, Gunter JL, O'Brien PC, Weigand SD, Knopman DS, et al. Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology. 2004; 62: 591-600. [CrossRef] [Google scholar] [PubMed]
- Aksu Y, Miller DJ, Kesidis G, Bigler DC and Yang QX. An MRI-derived definition of MCI-to-AD conversion for long-term, automatic prognosis of MCI patients. PLoS One. 2011; 6: 12. [CrossRef] [Google scholar] [PubMed]
- Carmichael O, McLaren DG, Tommet D, Mungas D and Jones RN. Coevolution of brain structures in amnestic mild cognitive impairment. Neuroimage. 2013; 66: 449-456. [CrossRef] [Google scholar] [PubMed]
- Devanand DP, Pradhaban G, Liu X, Khandji A, De Santi S, Segal S, et al. Hippocampal and entorhinal atrophy in mild cognitive impairment: prediction of Alzheimer disease. Neurology. 2007; 68: 828-836. [CrossRef] [Google scholar] [PubMed]
- Nestor SM, Gibson E, Gao FQ, Kiss A and Black SE. A direct morphometric comparison of five labeling protocols for multi-atlas driven automatic segmentation of the hippocampus in Alzheimer's disease. Neuroimage. 2013; 66: 50-70. [CrossRef] [Google scholar] [PubMed]
- Longstreth WT, Jr. Brain abnormalities in the elderly: frequency and predictors in the United States (the Cardiovascular Health Study). Cardiovascular Health Study Collaborative Research Group. J Neural Transm Suppl. 1998; 53: 9-16. [CrossRef] [Google scholar] [PubMed]
- Longstreth WT, Jr., Bernick C, Fitzpatrick A, Cushman M, Knepper L, Lima J, et al. Frequency and predictors of stroke death in 5,888 participants in the Cardiovascular Health Study. Neurology. 2001; 56: 368-375. [CrossRef] [Google scholar] [PubMed]
- Dufouil C, Chalmers J, Coskun O, Besancon V, Bousser MG, Guillon P, et al. Effects of blood pressure lowering on cerebral white matter hyperintensities in patients with stroke: the PROGRESS (Perindopril Protection Against Recurrent Stroke Study) Magnetic Resonance Imaging Substudy. Circulation. 2005; 112: 1644-1650. [CrossRef] [Google scholar] [PubMed]
- DeCarli C, Grady CL, Clark CM, Katz DA, Brady DR, Murphy DG, et al. Comparison of positron emission tomography, cognition, and brain volume in Alzheimer's disease with and without severe abnormalities of white matter. J Neurol Neurosurg Psychiatry. 1996; 60: 158-167. [CrossRef] [Google scholar] [PubMed]
- Longstreth WT, Jr., Manolio TA, Arnold A, Burke GL, Bryan N, Jungreis CA, et al. Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people. The Cardiovascular Health Study. Stroke. 1996; 27: 1274-1282. [CrossRef] [Google scholar] [PubMed]
- de Groot JC, de Leeuw FE, Oudkerk M, van Gijn J, Hofman A, Jolles J, et al. Cerebral white matter lesions and cognitive function: the Rotterdam Scan Study. Ann Neurol. 2000; 47: 145-151. [CrossRef] [Google scholar]
- Dawe RJ, Bennett DA, Schneider JA, Leurgans SE, Kotrotsou A, Boyle PA, et al. Ex vivo T2 relaxation: associations with age-related neuropathology and cognition. Neurobiol Aging. 2014; 35: 1549-1561. [CrossRef] [Google scholar] [PubMed]
- Prins ND, van Dijk EJ, den Heijer T, Vermeer SE, Koudstaal PJ, Oudkerk M, et al. Cerebral white matter lesions and the risk of dementia. Arch Neurol. 2004; 61: 1531-1534. [CrossRef] [Google scholar] [PubMed]
- Debette S and Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. Bmj. 2010; 341: 288. [CrossRef] [Google scholar] [PubMed]
- Thore CR, Anstrom JA, Moody DM, Challa VR, Marion MC and Brown WR. Morphometric analysis of arteriolar tortuosity in human cerebral white matter of preterm, young, and aged subjects. J Neuropathol Exp Neurol. 2007; 66: 337-345. [CrossRef] [Google scholar] [PubMed]
- Lo RY and Jagust WJ. Vascular burden and Alzheimer disease pathologic progression. Neurology. 2012; 79: 1349-1355. [CrossRef] [Google scholar] [PubMed]
- Provenzano FA, Muraskin J, Tosto G, Narkhede A, Wasserman BT, Griffith EY, et al. White matter hyperintensities and cerebral amyloidosis: necessary and sufficient for clinical expression of Alzheimer disease? JAMA Neurol. 2013; 70: 455-461. [CrossRef] [Google scholar] [PubMed]
- Toledo JB, Cairns NJ, Da X, Chen K, Carter D, Fleisher A, et al. Clinical and multimodal biomarker correlates of ADNI neuropathological findings. Acta Neuropathol Commun. 2013; 1: 2051-5960. [CrossRef] [Google scholar] [PubMed]
- Ferris SH, de Leon MJ, Wolf AP, Farkas T, Christman DR, Reisberg B, et al. Positron emission tomography in the study of aging and senile dementia. Neurobiol Aging. 1980; 1: 127-131. [CrossRef] [Google scholar] [PubMed]
- Minoshima S, Koeppe RA, Frey KA, Ishihara M and Kuhl DE. Stereotactic PET atlas of the human brain: aid for visual interpretation of functional brain images. J Nucl Med. 1994; 35: 949-954. [Google scholar]
- DeCarli CS AJ, Ball MJ, Kaye JA, Grady CL, Fewster P, Schapiro MB, et al. Postmortem regional neurofibrillary tangle densities, but not senile plaque densities, are related to regional cerebral metabolic rates for glucose during life in Alzheimer's disease. Neurodegeneration. 1992; 1: 113–121. [Google scholar]
- Wu X, Chen K, Yao L, Ayutyanont N, Langbaum JB, Fleisher A, et al. Assessing the reliability to detect cerebral hypometabolism in probable Alzheimer's disease and amnestic mild cognitive impairment. J Neurosci Methods. 2010; 192: 277-285. [CrossRef] [Google scholar] [PubMed]
- Langbaum JB, Chen K, Lee W, Reschke C, Bandy D, Fleisher AS, et al. Categorical and correlational analyses of baseline fluorodeoxyglucose positron emission tomography images from the Alzheimer's Disease Neuroimaging Initiative (ADNI). Neuroimage. 2009; 45: 1107-1116. [CrossRef] [Google scholar] [PubMed]
- Chen K, Langbaum JB, Fleisher AS, Ayutyanont N, Reschke C, Lee W, et al. Twelve-month metabolic declines in probable Alzheimer's disease and amnestic mild cognitive impairment assessed using an empirically pre-defined statistical region-of-interest: findings from the Alzheimer's Disease Neuroimaging Initiative. Neuroimage. 2010; 51: 654-664. [CrossRef] [Google scholar] [PubMed]
- Habeck C, Risacher S, Lee GJ, Glymour MM, Mormino E, Mukherjee S, et al. Relationship between baseline brain metabolism measured using [(1)(8)F]FDG PET and memory and executive function in prodromal and early Alzheimer's disease. Brain Imaging Behav. 2012; 6: 568-583. [CrossRef] [Google scholar] [PubMed]
- Ossenkoppele R, Prins ND, Pijnenburg YA, Lemstra AW, van der Flier WM, Adriaanse SF, et al. Impact of molecular imaging on the diagnostic process in a memory clinic. Alzheimers Dement. 2013; 9: 414-421. [CrossRef] [Google scholar] [PubMed]
- Elias A, Woodward M and Rowe CC. Management impact of FDG-PET in dementia: results from a tertiary center memory clinic. J Alzheimers Dis. 2014; 42: 885-892. [CrossRef] [Google scholar] [PubMed]
- Chen K, Ayutyanont N, Langbaum JB, Fleisher AS, Reschke C, Lee W, et al. Characterizing Alzheimer's disease using a hypometabolic convergence index. Neuroimage. 2011; 56: 52-60. [CrossRef] [Google scholar] [PubMed]
- Shaffer JL, Petrella JR, Sheldon FC, Choudhury KR, Calhoun VD, Coleman RE, et al. Predicting cognitive decline in subjects at risk for Alzheimer disease by using combined cerebrospinal fluid, MR imaging, and PET biomarkers. Radiology. 2013; 266: 583-591. [CrossRef] [Google scholar] [PubMed]
- Bohnen NI, Djang DS, Herholz K, Anzai Y and Minoshima S. Effectiveness and safety of 18F-FDG PET in the evaluation of dementia: a review of the recent literature. J Nucl Med. 2012; 53: 59-71. [CrossRef] [Google scholar] [PubMed]
- Mosconi L, De Santi S, Li J, Tsui WH, Li Y, Boppana M, et al. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol Aging. 2008; 29: 676-692. [CrossRef] [Google scholar] [PubMed]
- Fouquet M, Desgranges B, Landeau B, Duchesnay E, Mezenge F, de la Sayette V, et al. Longitudinal brain metabolic changes from amnestic mild cognitive impairment to Alzheimer's disease. Brain. 2009; 132: 2058-2067. [CrossRef] [Google scholar] [PubMed]
- Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, et al. Amyloid Deposition, Hypometabolism, and Longitudinal Cognitive Decline. Ann Neurol. 2012; 72: 578-86. [CrossRef] [Google scholar] [PubMed]
- Cohen AD, Price JC, Weissfeld LA, James J, Rosario BL, Bi W, et al. Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: an example of brain reserve. J Neurosci. 2009; 29: 14770-14778. [CrossRef] [Google scholar] [PubMed]
- Sabuncu MR, Desikan RS, Sepulcre J, Yeo BT, Liu H, Schmansky NJ, et al. The dynamics of cortical and hippocampal atrophy in Alzheimer disease. Arch Neurol. 2011; 68: 1040-1048. [CrossRef] [Google scholar] [PubMed]
- Oh H, Mormino EC, Madison C, Hayenga A, Smiljic A and Jagust WJ. beta-Amyloid affects frontal and posterior brain networks in normal aging. Neuroimage. 2011; 54: 1887-1895. [CrossRef] [Google scholar] [PubMed]
- Schott JM, Bartlett JW, Fox NC and Barnes J. Increased brain atrophy rates in cognitively normal older adults with low cerebrospinal fluid Abeta1-42. Ann Neurol. 2010; 68: 825-834. [CrossRef] [Google scholar] [PubMed]
- Chetelat G, Villemagne VL, Pike KE, Baron JC, Bourgeat P, Jones G, et al. Larger temporal volume in elderly with high versus low beta-amyloid deposition. Brain. 2010; 133: 3349-3358. [CrossRef] [Google scholar] [PubMed]
- Wirth M, Madison CM, Rabinovici GD, Oh H, Landau SM and Jagust WJ. Alzheimer's disease neurodegenerative biomarkers are associated with decreased cognitive function but not beta-amyloid in cognitively normal older individuals. J Neurosci. 2013; 33: 5553-5563. [CrossRef] [Google scholar] [PubMed]
- Doré V, Villemagne VL, Bourgeat P, Fripp J, Acosta O, Chetélat G, et al. CRoss-sectional and longitudinal analysis of the relationship between aβ deposition, cortical thickness, and memory in cognitively unimpaired individuals and in alzheimer disease. JAMA Neurology. 2013; 70: 903-911. [CrossRef] [Google scholar] [PubMed]
- Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex. 2009; 19: 497-510. [CrossRef] [Google scholar] [PubMed]
- Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, et al. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009; 65: 176-183. [CrossRef] [Google scholar] [PubMed]
- Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009; 132: 1310-1323. [CrossRef] [Google scholar] [PubMed]
- Storandt M, Mintun MA, Head D and Morris JC. Cognitive decline and brain volume loss as signatures of cerebral amyloid-beta peptide deposition identified with Pittsburgh compound B: cognitive decline associated with Abeta deposition. Arch Neurol. 2009; 66: 1476-1481. [CrossRef] [Google scholar] [PubMed]
- Chetelat G, La Joie R, Villain N, Perrotin A, de La Sayette V, Eustache F, et al. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer's disease. Neuroimage Clin. 2013; 2: 356-365. [CrossRef] [Google scholar] [PubMed]
- Villain N, Desgranges B, Viader F, de la Sayette V, Mezenge F, Landeau B, et al. Relationships between hippocampal atrophy, white matter disruption, and gray matter hypometabolism in Alzheimer's disease. J Neurosci. 2008; 28: 6174-6181. [CrossRef] [Google scholar] [PubMed]
- La Joie R, Perrotin A, Barre L, Hommet C, Mezenge F, Ibazizene M, et al. Region-specific hierarchy between atrophy, hypometabolism, and beta-amyloid (Abeta) load in Alzheimer's disease dementia. J Neurosci. 2012; 32: 16265-16273. [CrossRef] [Google scholar] [PubMed]
- Mormino EC, Betensky RA, Hedden T, Schultz AP, Amariglio RE, Rentz DM, et al. Synergistic effect of beta-amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA Neurol. 2014; 71: 1379-1385. [CrossRef] [Google scholar] [PubMed]
- Toledo JB, Bjerke M, Da X, Landau SM, Foster NL, Jagust W, et al. Nonlinear association between cerebrospinal fluid and florbetapir F-18 beta-Amyloid measures across the spectrum of Alzheimer disease. JAMA Neurol. 2015; 72: 571-581. [CrossRef] [Google scholar] [PubMed]
- Caroli A and Frisoni GB. The dynamics of Alzheimer's disease biomarkers in the Alzheimer's Disease Neuroimaging Initiative cohort. Neurobiol Aging. 2010; 31: 1263-1274. [CrossRef] [Google scholar] [PubMed]
- Walhovd KB, Fjell AM, Brewer J, McEvoy LK, Fennema-Notestine C, Hagler DJ, Jr., et al. Combining MR imaging, positron-emission tomography, and CSF biomarkers in the diagnosis and prognosis of Alzheimer disease. AJNR Am J Neuroradiol. 2010; 31: 347-354. [CrossRef] [Google scholar] [PubMed]
- Han SD, Gruhl J, Beckett L, Dodge HH, Stricker NH, Farias S, et al. Beta amyloid, tau, neuroimaging, and cognition: sequence modeling of biomarkers for Alzheimer's Disease. Brain Imaging Behav. 2012; 6: 610-620. [CrossRef] [Google scholar] [PubMed]
- Rabinovici GD, Rosen HJ, Alkalay A, Kornak J, Furst AJ, Agarwal N, et al. Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurology. 2011; 77: 2034-2042. [CrossRef] [Google scholar] [PubMed]
