

Redirecting Fetal Programming: Evidence of Interventions that May Be A Tool for Health
Camila Sandoval † ![]() , Colleen A Lambo †
, Colleen A Lambo † ![]() , Michael Carey Satterfield *
, Michael Carey Satterfield * ![]()
Physiology of Reproduction, Department of Animal Science, Texas A&M University, College Station, TX, USA
† These authors contributed equally to this work.
* Correspondence: Michael Carey Satterfield ![]()
Received: June 01, 2018 | Accepted: December 18, 2018 | Published: December 28, 2018
OBM Genetics 2018, Volume 2, Issue 4 doi: 10.21926/obm.genet.1804058
Academic Editors: Stéphane Viville and Marcel Mannens
Special Issue: Epigenetic Mechanisms in Health and Disease
Recommended citation: Sandoval C, Lambo CA, Satterfield MC. Redirecting Fetal Programming: Evidence of Interventions that May Be A Tool for Health. OBM Genetics 2018;2(4):058; doi:10.21926/obm.genet.1804058.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Understanding the pathophysiology of disease can be an essential step to determining where and how to intervene for preventive or corrective health. Intrauterine growth restriction (IUGR) has been defined in livestock species as impaired gestational development of a fetus or its parts [1]. This broad category of ailments described by low fetal weight is accompanied by susceptibility to adult onset of chronic disease through fetal programming of numerous organ systems and their functions. This concern affects humans and animals alike, representing a large sum of healthcare costs worldwide, and a substantial loss in animal agricultural yields. A gestating fetus requires an immense increase in nutrient delivery toward the end of pregnancy, which is achieved primarily through exponential increases in placental vascularization and uterine blood flow. An evaluation of the currently known mechanisms behind placental compromise can identify why some treatment interventions have successfully prevented IUGR, and identify new areas for experimentation. Of the many experimental models for IUGR, maternal nutrient restriction is a well-studied and minimally invasive tool with real-world application to famine and grazing livestock. Nutrient restriction in the sheep model has demonstrated that compromised fetal development can be improved through treatment with sildenafil citrate, and that reduced populations of amino acid transporters can be supported by increasing maternal circulating concentrations of specific amino acids. In addition, a number of prenatal treatments such as amino acid, selenium supplementation, and realimentation have been successful at or have the potential to prevent the IUGR phenotype or the metabolic alterations leading to higher risk of disease. While the thrifty phenotype has long been a cause for health issues worldwide, available research from the nutritionally restricted pregnant ewe offers hope for identifying treatment interventions to redirect or reverse concerning developmental trajectories.
Keywords
IUGR; nutrient restriction; sheep; placental insufficiency; epigenetics; thrifty phenotype
1. Introduction
While our genetic codes have long been considered the primary marker for what attributes we are born with, a recent flurry of research has uncovered the concept of fetal programming. Historically, the ‘nature vs nurture’ theory considered our DNA in comparison to life events occurring after birth as the determinants of what we became. Fetal programming, however, involves modifications made to the DNA structure in-utero through epigenetic modifications, which results in permanent alterations in the expression of genes in response to our environment. A wide variety of adverse uterine environments have been studied; from fetal alcohol syndrome, to nutritional extremes, hypoxia, hyperthermia, and exposure to environmental toxins. Under adverse conditions, the gestating female and the fetus will attempt to prepare for the harsh environment she is experiencing. Unfortunately, these adaptations can be problematic if there is an environmental mismatch, and the conditions the infant was prepared for do not occur. Offspring that experience this mismatch will be at even greater risk for long term health implications such as increased risk for mortality, diabetes, obesity, cardiovascular disease, and metabolic syndrome [2].
The first epidemiological studies to recognize this phenomenon were described by Dr. David Barker following his evaluations of adults that were carried during periods of famine. He proposed the thrifty phenotype hypothesis, which states that intrauterine growth restriction (IUGR), and low birth weights impart a higher incidence of non-insulin dependent diabetes, hypertension, and coronary heart disease in adults [3,4,5]. Nutritional restriction remains relevant today as famine continues to be a worldwide problem, with an estimated 815 million undernourished people around the world [6]. As we look forward, the agricultural demands to feed a rapidly growing population on fewer natural resources is an eminent problem to be addressed. Failure to adequately do so will only exacerbate the world hunger problem. The research necessary to elucidate the gross and molecular responses to nutrient restriction during pregnancy has been carried out in a variety of animal species, as well as retrospectively in humans. Of these, the sheep model has proven to be of incredible value to both the agricultural and human health industries. The hemochorial human placenta is most similar to the rodent model, but rodent gestation is substantially shorter, and young are born precocial, which limits the ability to introduce and monitor insults for developmental studies, particularly insults occurring in late gestation (Figure 1). The sheep has long been an established model for investigations related to fetal development and physiology due to their comparable level of organ development at birth, similar size at birth, non-litter bearing status, capacity for surgical manipulation, and blood sampling capabilities. The pregnant sheep also serves as a valuable model for itself and other ruminants for meat and milk production throughout the world.
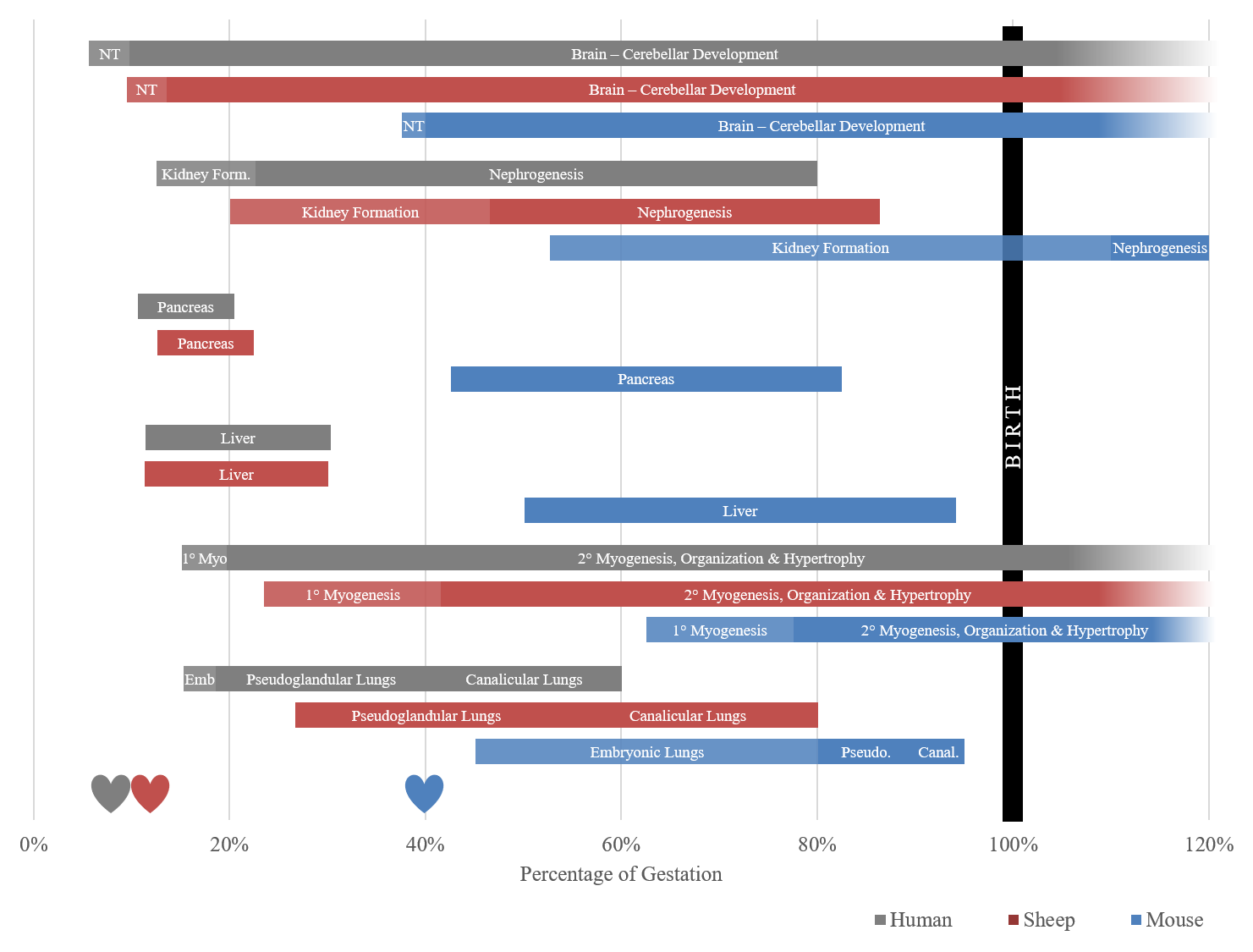
Figure 1 Timeline of organogenesis based on percent of gestation in the human, sheep and mouse. Development windows represent a time when organs are most susceptible to insult, albeit they can be compromised outside of these windows. The sheep timeline is a closer approximation for the human timeline than the mouse, which is born precocial, and is 40% through gestation when the neural tube (NT) forms. Hearts in the figure represent detectable presence of a heartbeat. 1° Myo refers to the period of primary myogenesis of skeletal muscle, Emb is the phase of embryonic lung development, compared to pseudoglandular lung (Pseudo.) and canalicular lungs (Canal.) [7,8,9,10,11,12,13,14,15,16,17,18,19].
Sheep nutrient restriction models (NR; 70% - moderate; 50% - severe, 30-40% - extreme) have been implemented to study the impacts of famine on fetuses. Offspring from restricted ewes are likely to be at higher risk for postnatal diseases, and experience IUGR, which, in livestock, is broadly defined as impaired growth of the fetus or its organs [20,21]. Small fetal size is not itself pathological, yet the growth restriction is inextricably linked to the additional negative sequela. Research in sheep has shown that IUGR results from impaired placental development, which restricts subsequent fetal development. By determining the mechanisms through which placental development or fetal nutrient delivery has been compromised, studies [22,23] have indicated that treatment with pharmaceuticals or feed supplements may be able to protect the placenta and/or fetus by preventing the response to the insult from occurring. Therapies that can prevent the lifelong impacts of IUGR, or otherwise steer fetal programming toward a healthier offspring are valuable to identify. This review will focus on identifying the physiology causing fetal programming following low nutrient intake in gestating ewes, as well as the subsequent phenotypic responses seen in the offspring, and interventions that have shown potential for mitigating the programming concerns (Table 1).
Table 1 Summary of selected studies which demonstrate opportunities for physiological interventions to halt the trajectory toward a thrifty phenotype in the nutrient restricted sheep model.
|
Item |
Tissue |
Treatment |
Gestational Day |
Effect |
Reference |
|
Placenta |
Placenta |
70% NR |
0 to term |
-20% lower maternal caruncular capillary area density |
[24] |
|
Realiment |
90 to 130 |
-Did not improve capillary area density or IUGR |
|||
|
30-40% NR |
28 to 108 |
-Decreased uterine blood flow – doppler -IUGR, hypoglycemic |
[25] |
||
|
30-40% NR |
120 to 143 |
-Uterine bloodflow reduced 30% -IUGR, hypoglycemic -Placental mass unchanged |
[26] |
||
|
50% NR |
28 to 78 28 to 135 |
-Reduced AA & polyamines in fetal blood & placental fluids -Greater reduction in AA & polyamines |
[27] |
||
|
Realiment |
78 to 135 |
-AA concentration increased to control levels |
|||
|
50% NR |
28 to 115 |
-Decreased fetal weight |
[22] |
||
|
Viagra |
28 to 115 |
-Increased fetal weight -Increased polyamines & total AA in placental fluids |
|||
|
50% NR |
28 to term |
-Decreased birthweight 23% -Reduced concentration of 20 amino acids |
[23] |
||
|
IV Arginine |
60 to term |
-Increased birthweight 21% |
|||
|
Glucose/Insulin Metabolism |
Skeletal Muscle |
50% NR |
28 to 78 |
-Increased proportion of type II myofibers -Low GLUT4 content |
[28] |
|
50% NR |
85 to 115 |
-Decreased muscle mass |
[29] |
||
|
50% NR |
28 to 78 |
-Decreased phosphorylated mTOR and ribosomal protein S6 |
[30] |
||
|
Pancreas |
50% NR |
28 to 78 |
-Increased insulin secretion -Low insulin sensitivity |
[31] |
|
|
50% NR |
110 to term |
-Insulin resistance -Glucose intolerance |
[32] |
||
|
50% NR |
35 to 125 |
-Decreased pancreas weight |
[33] |
||
|
IV Arginine |
100 to 125 |
-Increased pancreas weight by 32% |
|||
|
60% NR |
50 to term |
-Decreased insulin secretion |
[34] |
||
|
Selenium |
0 to term |
-Increased insulin secretion |
|||
|
70% NR |
26 to term |
-Decreased pancreas weight |
[35] |
||
|
Liver |
50% NR |
28 to 78 |
-Increased relative fetal liver weight |
[36] |
|
|
50% NR |
0 to 95 |
-Decreased liver weight in aged offspring -Upregulation of the apoptotic factor Bax -Downregulation of GH, Prolactin, and IGFII receptors |
[37] |
||
|
50% NR Realiment Obesogenic |
30 to 80 80 to term Wean to 1yr |
-Increased hepatic triglyceride content -Increased PPARƴ and PGC1α mRNA |
[38] |
||
|
50% NR Realiment
|
31 to 100 100 to term
|
-Increased PEPCK mRNA & protein in aged male offspring -Hypomethylation of glucocorticoid receptor in aged male -Upregulated glucocorticoid receptor mRNA in aged male -Low hepatic weight in aged male offspring |
[39] Exp. 2 |
||
|
50% NR Realiment |
0 to 30 30 to term |
-Increased PEPCK mRNA & protein in aged male offspring -Upregulated glucocorticoid receptor mRNA in aged male |
[39] Exp. 1 |
||
|
Cardiovascular/Hypertension |
Heart |
70% NR |
26 to term |
-Decreased heart weight |
[35] |
|
50% NR |
28 to 78 |
-Increased ventricular thickness |
[36] |
||
|
50% NR |
115 to term |
-Increased hypertrophic signalling (CAMKII, IGF2, IGF2R) -Ventricle interstitial fibrosis and collagen deposition -Hypertension |
[40] |
||
|
Blood Vessels |
50% NR |
50 to 130 |
-Decreased vasodilation capacity in coronary arteries |
[41] |
|
|
Kidney |
50% NR |
28 to 78 |
-Decreased glomeruli number -Increased mRNA expression of ACE and AT2 -Hypertension |
[42] |
|
|
Protein restriction |
0 to 65 |
-Impaired renal microvasculature development -Impaired fetal ornithine cycle, decreased polyamine synthesis |
[43] |
||
|
Protein restriction |
0 to 65 |
-Impaired nephrogenic potential -Upregulated apoptosis, downregulated angiogenesis |
[44] |
||
|
Obesity |
White adipose (WAT) |
50% NR |
28 to 78 |
-Increased WAT tissue weight (adult peri-renal & pelvic depots) |
[45] |
|
Appetite Control |
70% NR |
6 to 130 |
-Decreased leptin and increased neuropeptide Y expression |
[46] |
|
|
Realiment |
90 to 130 |
-Normalized leptin mRNA expression |
|||
|
Brown adipose (BAT) |
50% NR |
35 to 125 |
-Decreased BAT weight (peri-renal depot) |
[33] |
|
|
IV arginine |
100 to 125 |
-Increased BAT weight by 48% |
* Shaded boxes indicate attempted programming counter-treatments.
2. Why Epigenetics?
2.1 Preparing the Offspring for An Anticipated Environment
It has been suggested that when the maternal organism is under adverse environmental conditions, the dam will perceive these environmental signals and respond through cues, and the fetus will respond accordingly to be better adapted for the anticipated postnatal habitat [47]. The ability to adapt during early life development is an important biologically conserved phenomenon, termed developmental plasticity. Developmental plasticity allows for fine-tuned regulation of critical physiological set points in response to the specific environment that the fetus is developing in, through non-genomic mechanisms. When the anticipated environment matches the conditions that the offspring faces during postnatal life, the adaptation will favor survival of those individuals. However, a mismatch will produce offspring that are adapted for the wrong environment. This can lead to increased morbidity and mortality [48]. Maternal nutrient restriction can be an example of a ‘mismatch’ if the offspring is exposed to nutritional excess after birth [38]. In these instances, the anticipation for an adverse postnatal environment will induce a fetal adaptation for survival in nutrient scarcity, which will also make it more likely to develop diseases under normal or excess nutritional levels. This has been described by Barker and Hales as the thrifty phenotype hypothesis, which suggests that the fetus will adapt to maternal malnutrition by programming a phenotype that is prepared to “save” and “store” nutrients rather than immediately using them [3]. Nevertheless, under postnatal nutritional excess, the offspring will be more prone to develop diseases as a consequence of a prenatal stimuli; this is an example of “developmental origins of health and disease” (DOHaD), which refers to environmental conditions in early life that will increase the risk for disease in postnatal life [49].
2.2 Molecular Pathways for Programming: Epigenetics
When malnourished during pregnancy, the dam will mobilize her own reserves in an attempt to promote offspring survival, and she will lose weight as the nutritional demand to support fetal growth becomes greater. This response may not successfully compensate for the lack of nutrients, and placental development could be affected by maternal undernutrition, thus limiting nutrient delivery to the fetus [50]. In order to adapt to this adverse environment and survive, the fetal response to nutrient scarcity in utero needs to be rapid and mediated by a mechanism that can translate the ambient signals into effects in-line with the rapid pace. Modifications of the DNA sequence would be stable, but slow. Through epigenetics, however, gene expression can be modulated to induce phenotypic effects, without editing DNA sequence [51]. Epigenetics can be understood as biochemical modifications to the non-genomic structure of the chromosomal material that control the expression of genes by affecting their promotor region, but do not alter the genetic code. This leads to the existence of a common genotype in all the cells of an organism, but infinite possible combinations of epigenomes. The two primary epigenetic modifications described in literature to date are DNA methylation and histone modifications [52].
DNA methylation. In mammals, the addition of methyl groups to cytosine residues in CpG islands within the DNA is the most characterized epigenetic modification [53]. CpG islands are regions in the genome that are enriched in cytosine and guanine, and are often located in the promotor regions of genes, which are in control of gene transcription. Methylation in these regions will lead to transcriptional inhibition and further downregulation of gene expression [54]. In the context of maternal undernutrition, methylation processes are particularly susceptible because the availability of methyl groups is controlled by nutritional factors. Among them, the amino acids glycine, methionine, serine, and histidine play a role by regulating one-carbon metabolism, which determines the availability of S-adenosyl-methionine, a major methyl donor in the organism (Figure 2). Histidine and methionine are classified as essential amino acids, which means that the mammalian organism cannot synthesize it, thus it has to be obtained exclusively from the diet. Glycine, and serine are classified as non-essential amino acids, which means that the organism can synthesize them. Nevertheless, the concept of conditionally essential amino acid states that even when an amino acid can be synthesized in the organism, this might not be enough under highly demanding physiological situations, such as pregnancy, and may become even more critical when the dam is malnourished during pregnancy [55]. Other nutrients such as vitamin b12 and folate play essential roles in the control of one-carbon metabolism as well, which will be limiting in NR conditions and can have an effect on methyl donor availability [56]. This has been validated using a sheep model of dams fed low vitamin B12, folate, and methionine from 8 weeks prior to 6 days following conception, which altered DNA methylation and led to insulin resistance, hypertension, increased body fat, and decreased lean mass in 12 month-old male offspring [57].
Histone modifications. Histone modifications include acetylation, methylation, phosphorylation, ubiquitination, sumoylation, carbonylation, and poly(ADP-ribosyl)ation [53]. In general, these modifications will influence how tightly packaged the histone-DNA is; the tighter, the more likely it is to restrict translation [58]. As with DNA methylation, histone methylation also depends on nutritional factors that regulate the availability of methyl groups in the organism through one carbon metabolism (Figure 2). Methylation of histones can either induce or repress gene expression depending on the location of the modification [59]. It is not yet known how environmental stimuli induce histone modifications other than methylation [53].
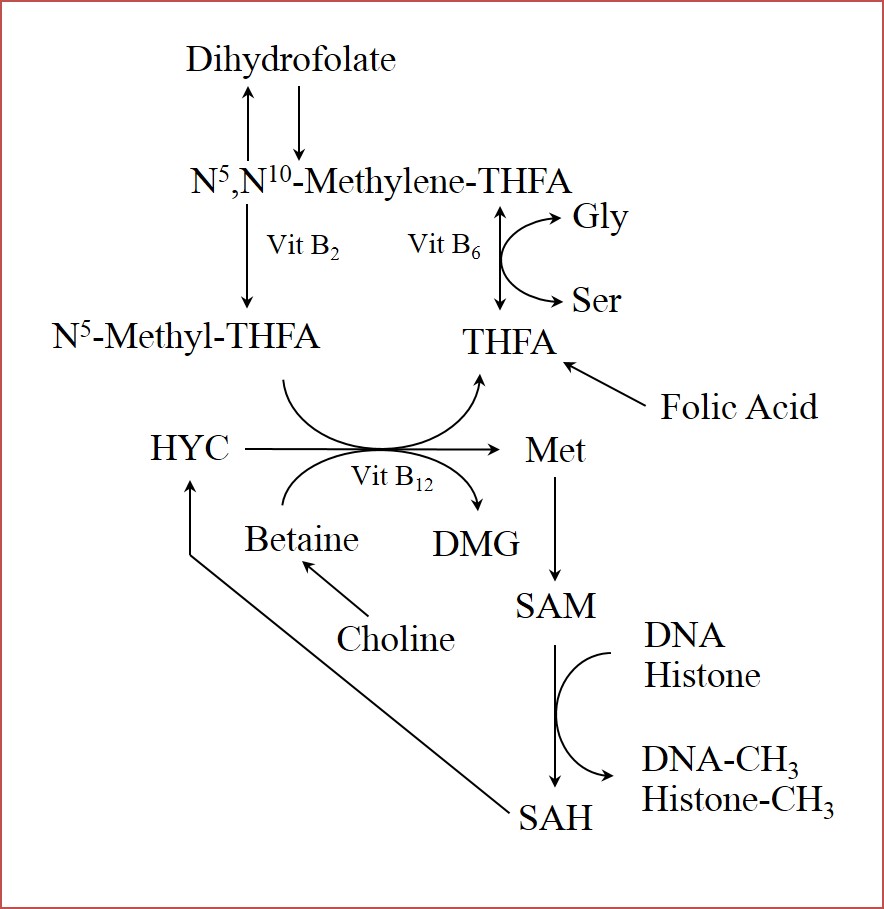
Figure 2 Role of nutrients in regulation of one-carbon-unit metabolism. Amino acids glycine, serine, and methionine; and vitamins (B2, B6, B12, and Folic Acid) are involved in the provision of methyl groups, which are required for methylation to either DNA or histones. Methionine acts as precursor of S-adenosylmethionine, which is a major methyl group donor in the organism. Conversion of S-adenosylmethionine to S-adenosylhomocysteine releases a methyl group, which is used as substrate in DNA, and histone methylation. Gly (glycine); Ser (serine); Met (methionine); SAM (S-adenosylmethionine); SAH (S-adenosylhomocysteine); DMG (dimethylglycine); HYC (homocysteine); THFA (tetrahydrofolate).
The majority of research studying epigenetic modifications in response to maternal nutrient restriction has focused on DNA methylations [53]. It has been suggested that different epigenetic modifications are acting together [60] to control gene expression, induce a specific phenotype in the offspring, and lead to a biological effect that could be evident either at birth or later in postnatal life.
3. Physiologic and Phenotypic Effects of Maternal Undernutrition
3.1 Effects on Placental Development
The trajectory and competence of placental growth, development, and vascularization is essential to subsequent successful fetal development and health. In the first half of pregnancy the placenta undergoes proliferative growth, while placental vasculature continues to undergo significant development in the 2nd half of gestation, in order to support the exponential fetal growth phase [24]. The ovine placenta is cotyledonary in shape and has 60-120 discrete regions of intensive maternal (caruncle) and fetal (cotyledon) epithelial cell interdigitation, termed placentomes, to provide sufficient surface area for efficient nutrient, gas, and waste exchange. Placentomes are highly vascularized regions of the placenta with over 84% of maternal uterine blood flow and 94% of fetal blood flow traveling through these structures [61]. From gestational day (GD) 90 – 130, capillary area density increases approximately 2-fold in fetal cotyledons, and 1.5-fold in maternal caruncles [24,62]. Gestational day 90 represents a high phase of placental growth and the start of exponential vascular development within the cotyledon, while GD130 is considered a time of maximal placental vascularization, blood flow and nutrient transport [24]. The majority of impairments in fetal growth begin with compromised development of the placenta, its vascularization, or nutrient transport systems.
Restricted development of the placenta directly impacts the development of the fetus. Uterine blood flow and placental vasculature seem to be the most susceptible to insult, and make the largest impact, especially during late gestation. In a moderate NR study on adolescent ewes, placental gross morphology, cellular proliferation & mass did not change in the NR group, but a 20% reduction was seen in maternal caruncular capillary area density, which was greater than the observed percent reduction in fetal weights. In a subgroup where realimentation was attempted after GD90, the capillary density and fetal weight reduction did not improve [24]. In extremely NR ewes, placental masses were unchanged, while uterine blood flow was reduced by 30%, fetal weights were reduced, and fetuses were hypoglycemic [25,26]. While placental mass can be reduced in NR models, these studies indicate that the reduction in vasculature is the more important factor in restricting nutrient delivery. The failure of realimentation to correct vascular deficiency implies that the developmental trajectory was established prior to the correction at GD90. Additionally, Lang et al. mechanically restricted uterine blood flow, and showed a greater compromise to the fetus with a greater restriction in blood flow [63]. The reduction of placental vascular development through NR diets is a predictable and important change that greatly impacts subsequent fetal development and health. With consideration for this pathophysiology, treatment with sildenafil citrate (Viagra) was considered as a potent vasodilator. Administration of Viagra from GD28 to GD115 in NR sheep increased fetal weight, preventing the IUGR condition in the restricted group. These effects were seen with an increase in polyamines and total amino acids in amniotic and allantoic fluids [22], which may have been caused by enhanced delivery due to increased uterine blood flow. This discovery was the first direct evidence that sildenafil citrate could be used as therapy to enhance nutrient delivery to fetuses and prevent IUGR. Human trials with Viagra, however, have been halted after a failure to see comparable improvements in placental blood flow, and a potentially coincidental occurrence of lung morbidity and infant mortality in one study group [64].
Amino acids, which are essential protein building blocks for a growing fetus, and may act as regulators of placental and fetal growth [65] must be actively transported across fetal membranes. The described increase in capillary area density & surface area for exchange in a healthy fetus is not sufficient to account for the vast increase in amino acid demand during exponential fetal growth. The density and type of transporters and circulating maternal concentrations of amino acids increase to meet the demands [65,66]. Kwon et al. [27] showed that circulating total amino acids & polyamines were reduced in NR ewes by day 78 and remained low on day 135 in maternal & fetal plasma and fetal fluids. Realimentation in a separate group starting on day 78 was able to correct the reductions. Supplementation of single amino acids is likely to increase fetal levels, but amino acid cocktails will have mixed results due to transporter competition [65,66]. Intravenous administration of arginine alone to NR dams from GD60 to term successfully enhanced birth weight by 21%, potentially protecting the fetuses from IUGR [23]. Not only is arginine a valuable amino acid, it supports angiogenesis and may have contributed to fetal growth through increased fetal delivery of arginine, as well as increased delivery of nutrients in general due to the vascular support, through its metabolite, nitric oxide.
3.2 Effects on Fetal Metabolism and Postnatal Disease
Offspring born to nutrient restricted females are under a higher risk of disease; primarily hypertension, obesity, diabetes, insulin resistance, heart disease, and metabolic syndrome in adulthood [20]. Under maternal NR conditions, organs in the fetus will have different susceptibility to damage caused by prioritization of the nutrient supply towards more vital organs, such as the brain and heart; while “non-vital” organs, such as skeletal muscle, and the pancreas are more likely to experience impaired development because of lower nutrient priority [67]. The type and severity of stimuli, as well as the stage of gestation are important to consider in relation to fetal programming potential. A “window of sensitivity” describes when different organs will be more or less susceptible to programming depending on the stage of organogenesis or development when the insult occurred. Therefore, an identical insult (i.e. nutritional restriction) may lead to different postnatal phenotypes depending upon the period of gestation that the insult was applied. Indeed, apparent conflicting results observed in scientific studies of fetal programming are often due to differences in timing and/or duration of the insult.
Effects on Glucose/Insulin Metabolism. It is traditionally accepted that maternal NR induces IUGR, and this condition usually leads to decreased muscle mass [29,68]. Skeletal muscle represents 40% of total body mass [69], and accounts for 80% of insulin-mediated glucose disposal in the organism, so that an alteration in muscle mass or metabolism will impact whole-body glucose metabolism [67]. There are multiple pathways through which NR can decrease fetal muscle mass, the most studied of which is mTOR signaling pathway, which acts as an activator of protein translation and deposition. The mTOR signaling pathway is stimulated by insulin, glucose, IGF-1, and amino acids such as leucine, arginine, and glutamine [70], and is downregulated as a consequence of maternal undernutrition [71]. As an example, Zhu et al. [30] found no differences in total mTOR protein, but a decrease in phosphorylated mTOR in fetal skeletal muscle from ewes restricted between GD 28 to 78. A phosphorylated form of ribosomal protein S6, a downstream target of mTOR, was also decreased. Downregulation on mTOR signalling was indicative of lower protein translation in fetal skeletal muscle in response to maternal nutrient restriction.
Myofiber composition is also relevant for muscle metabolism and whole-body insulin sensitivity. Type I myofibers are primarily oxidative, and more sensitive to insulin than type II, which are primarily glycolytic [72]. Data from sheep models of severe maternal NR in early pregnancy showed increased proportion of type II myofibers, and lower content of glucose transporter 4 (GLUT4), the major insulin-stimulated glucose transporter in skeletal muscle, in 8 month-old lambs [28].
Maternal NR has also been shown to affect development of the pancreas and insulin secretion in the offspring. A severe NR throughout pregnancy resulted in decreased fetal pancreas weights in sheep [35]. Severe NR during early pregnancy has been related to increased insulin secretion but lower insulin sensitivity in adult offspring [31]. Glucose intolerance and insulin resistance were identified in young offspring using a sheep model of NR in late pregnancy, but not when the restriction occurred in early pregnancy. Decreased GLUT4 expression in adipose tissue was found as a primary cause of glucose metabolism dysregulation in those animals [32]. In addition to increasing fetal weights as discussed previously, IV arginine administration from GD100 to GD125 increased pancreatic weights by 32% in a severe NR model [33]. It is anticipated that this will have a beneficial impact on amelioration of postnatal glucose metabolism dysregulation, induced as a consequence of maternal NR. Additional research has demonstrated that selenium supplementation throughout pregnancy increases insulin release in 107 day-old lambs born to 60% NR dams [34]. This intervention could more favorably program glucose metabolism in aged offspring, yet this remains to be demonstrated.
The liver is also involved in glucose metabolism as it responds to hypoglycemia by gluconeogenesis. Severe undernutrition from GD 28 to 78 led to increased hepatic weight per unit of fetal weight at 78 days of gestational age [36]. By contrast, Hyatt et al. [37] found disproportionately smaller livers and upregulation in hepatic apoptotic factors. Liver was also found to be affected in 1 year-old offspring fed an obesogenic diet, after a prenatal severe NR from GD30 to 80. Particularly, hepatic triglyceride content and mRNA expression of peroxisome proliferator-activated receptor ƴ (PPARƴ) & PGC1α (a coactivator of PPARƴ) were increased [38].
Hepatic PEPCK gene and protein expression was increased in 6 year-old offspring fed 100% of their nutritional requirements after a prenatal severe NR from GD 28 to 78 [31]. PEPCK is a rate-limiting enzyme in gluconeogenesis, therefore its upregulation may be an indicator of nutritional programming towards upregulated endogenous glucose production. Chadio et al. [39] also found increased mRNA and protein expression of PEPCK in 10-month-old males born to ewes under severe NR either from GD0 to GD30, or GD31 to GD100. This study also showed hypomethylation in liver glucocorticoid receptor (GR) gene promoter in aged male offspring of NR ewes between GD31 and GD100, while a tendency was found in males born to NR sheep from GD0 to GD30. Upregulated GR mRNA expression was found in both restricted groups. GR is a transcription factor known to activate the PEPCK gene [73] and may explain the previously described increase in PEPCK mRNA.
Effects on Cardiovascular Development and Hypertension. The initial epidemiological correlation between maternal undernutrition and increased risk for cardiovascular disease in offspring was established by epidemiologist Dr. David Barker [74]. Using the sheep model, several studies have attempted to explain the underlying causes of this predisposition. Decreased fetal heart weight has been demonstrated in response to maternal NR throughout pregnancy [35]. Increased ventricular wall thickness, or cardiac hypertrophy has been observed at GD78 in offspring from dams restricted from early to mid-gestation [36]. This effect was later associated with increased expression of cardiac insulin-like growth factor receptors [75]. Similarly, an upregulation in hypertrophic signalling mediated CAMKII activation by increased expression of IGF2 and IGF2R was observed in sheep fetuses from dams undernourished in late gestation. Interstitial fibrosis and increased collagen deposition in the right ventricle and increased arterial blood pressure by GD145 were also seen [40]. In addition to cardiac abnormalities, which predispose to cardiovascular sequela in adults, altered development of vasculature have been associated as a contributor to cardiovascular disease and hypertension. As an example, it has been shown that coronary arteries of sheep fetuses from NR dams during late pregnancy are less prone to vasodilate because of impairment in the endothelium-derived hyperpolarizing factor-like pathway [41].
Effects on Renal Tissue and Clearance. Impairment of kidney development plays a role in the onset of hypertension following gestational restriction. This has been associated with decreased nephron and glomeruli development [76]. As an example, severe NR from GD 28 to 78 induced increased mean arterial pressure, a decreased glomeruli number and an increased expression of angiotensin converting enzyme and angiotensin II receptors in the offspring by 9 months of age [42]. Dunford et al. [43] found that a maternal protein restricted but isocaloric diet impairs the ornithine cycle in the sheep fetus, decreasing the ability to synthesize polyamines. This was suggested as an underlying cause of impaired renal microvasculature development, mostly in the nephrogenic areas, leading to low nephron endowment. This is supported by results of Lloyd et al. [44] who demonstrated that maternal protein and energy malnutrition leads to impaired nephrogenic potential associated with upregulated apoptosis and downregulated angiogenesis.
Effects on Brown Adipose Tissue Development and Obesity. There is a higher risk of developing obesity in offspring from NR dams [4]. Severe NR during early to mid-pregnancy in sheep led to increased white adipose tissue deposition in peri-renal and pelvic depots in 4 month old offspring [45]. Programmed hyperphagia will contribute to the onset of obesity by providing excess nutrients to the organism. One signalling molecule that controls satiety in mammals is leptin. Moderate NR throughout gestation decreased mRNA expression of leptin and increased orexigenic factor neuropeptide Y in sheep fetuses by GD130. Importantly, maternal realimentation in late gestation was successful in increasing leptin mRNA expression to control levels [46] which suggests that restoration of appropriate nutritional levels can at least partly serve as a possible intervention to mitigate hyperphagia in postnatal life.
Brown adipose tissue (BAT) is essential in non-shivering thermogenesis, which plays a fundamental role in neonatal survival. Heat is released as a result of fatty acids and glucose oxidation, because of expression of uncoupling protein 1 (UCP-1) in BAT mitochondria, which uncouples the electron transport chain with ATP synthesis, and energy is dissipated as heat instead [77]. In addition to its role in neonatal survival, BAT plays a central role in energy balance and glucose metabolism, because of its capacity to burn fatty acids and glucose [78]. Studies in humans have demonstrated the presence of functional BAT in adults [79,80], which was previously considered to be metabolically inactive; this has redirected attention to this tissue as a potential target to prevent obesity and enhance metabolic profiles [81]. Evidence to support this comes from the finding that obese adult humans have lower BAT activity, measured as cold-induced thermogenesis, than lean individuals [82]. In addition, stimulation of BAT activity is negatively correlated with body fat mass in adult humans [83].
Following a severe NR treatment from GD35 to GD125 in the sheep, fetal BAT weight decreased. Intravenous IV arginine supplementation from GD100 to GD125 increased BAT weight by 48% [33]. Results have also demonstrated a beneficial effect from arginine on BAT of sheep offspring from obese dams, reinforcing the potential therapeutic effects of arginine in the control of programmed obesity [84]. A study using brown adipocyte precursors isolated from fetal lambs demonstrated that arginine enhances BAT growth, inducing protein synthesis and cell growth mediated by upregulation of the mTOR signalling pathway [85].
Collectively these results indicate that arginine supplementation is a promising intervention strategy to mitigate the risk of postnatal obesity in IUGR fetuses. Further research will determine if results will remain in aged offspring. A recent discovery that sternal fat in adult sheep conserves UCP-1 expression and heat production capacity supports the use of the sheep model to study BATs role in preventing obesity in aged offspring [86]. This makes the sheep a suitable model to evaluate prenatal interventions to prevent postnatal obesity by interventions that program BAT growth and metabolism.
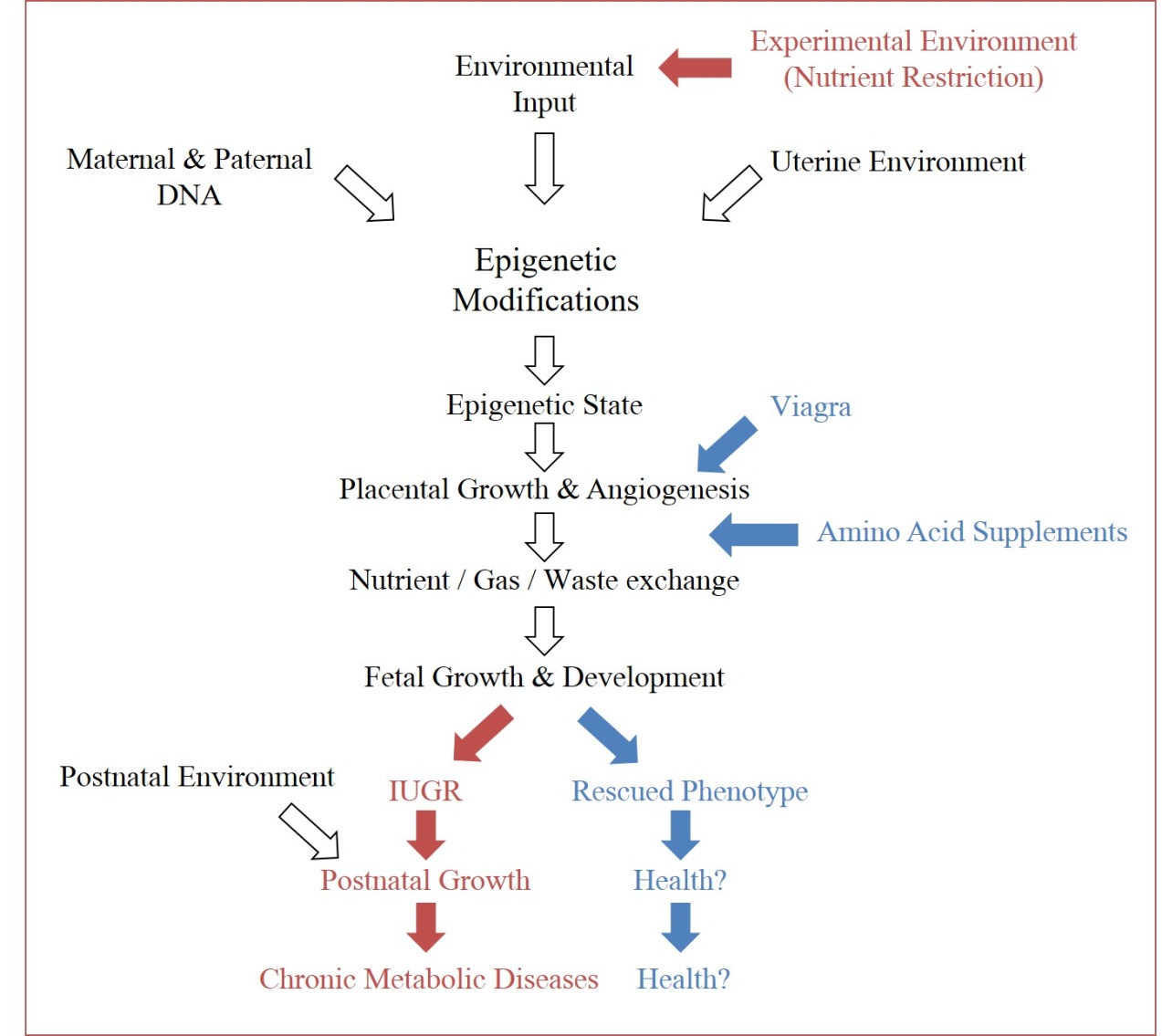
Figure 3 Experimental opportunities in epigenetics. Through recognition of environmental impacts on epigenetic and subsequent phenotypic outputs, experimental conditions can force the ‘thrifty phenotype hypothesis’ (maroon arrows), in order to study the physiological cascade which connects the two (open arrows). Subsequently, intervention opportunities to redirect offspring from negative phenotypes can be identified along the pathway (examples in blue arrows).
4. Discussion
Gestational insults, specifically restriction of nutrients during gestation, can impact fetal development through impaired placental growth and subsequently impaired fetal development. The uterine environment imparts adaptive programming to the epigenome of the fetus, which modulates expression of the inherited genetic code (Figure 3). These modifications are meant to prepare the fetus for a harsh environment, and can be beneficial if those conditions are observed. If there is a mismatch between the anticipated environment and the true environment, these programmed adaptations can prove harmful. With better awareness for the physiological cascade that induces these modifications, it may not only be possible to intervene during a known insult to protect the fetus from future anticipated disease susceptibility, but also to use these programming designs to support fetuses compromised by an array of other challenges, which may share similar pathways to those discovered. Unlike genetic information, epigenetic events are dynamic in their response to endogenous and exogenous signals, meaning they have the potential to be redirected [87]. This plasticity allows us to consider the redirection of the programmed hallmarks that maternal undernutrition induces in offspring, which can lead to the IUGR phenotype, and metabolic disorders.
5. Conclusions
For the past decades, scientists studying epigenetics and fetal programming have focused mostly on describing how stimuli in early life impact development, and how mismatches with the postnatal environment increase the risk for disease (DoHAD paradigm). There has been great progress in this field, however, we still do not fully understand precisely how different diseases are programmed. Nevertheless, there is a need to take the next step and direct scientific efforts towards answering a different question: Can we prevent disease by proscriptively programming for health during prenatal life? Increasing knowledge from this scope could change our view of medicine, from primarily postnatal and curative to prenatal and preventive.
Author Contributions
Each author participated in the research and writing of this article.
Competing Interests
The authors have declared that no competing interests exist.
References
- Reynolds LP, Caton JS, Redmer DA, Grazul-Bilska AT, Vonnahme KA, Borowicz PP, et al. Evidence for altered placental blood flow and vascularity in compromised pregnancies. J Physiol. 2006; 572: 51–58. [CrossRef] [Google scholar] [PubMed]
- Gluckman P, Hanson M, Buklijas T. A conceptual framework for the developmental origins of health and disease. J Dev Orig Health Dis. 2010; 1: 6–18. [CrossRef] [Google scholar] [PubMed]
- Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1992; 35: 595–601. [CrossRef] [Google scholar] [PubMed]
- Ravelli AC, van der Meulen JH, Osmond C, Barker DJ, Bleker OP. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr. 1999; 70: 811–816. [CrossRef] [Google scholar] [PubMed]
- Barker DJ. Fetal growth and adult disease. BJOG. 1992; 99: 275–276. [CrossRef] [Google scholar] [PubMed]
- FAO. The state of food security and nutrition in the world 2017: Building resilience for peace and food security 2017. [Google scholar]
- Ji M, Bai C, Li L, Fan Y, Ma C, Li X, et al. Biological characterization of sheep kidney-derived mesenchymal stem cells. Exp Ther Med. 2016; 12: 3963–3971. [CrossRef] [Google scholar] [PubMed]
- McMahon AP. Development of the mammalian kidney. Curr Top Dev Biol. 2016; 117: 31–64. [CrossRef] [Google scholar] [PubMed]
- Wilson SJ, McEwan JC, Sheard PW, Harris AJ. Early stages of myogenesis in a large mammal: formation of successive generations of myotubes in sheep tibialis cranialis muscle. J Muscle Res Cell Motil. 1992; 13: 534–550. [CrossRef] [Google scholar] [PubMed]
- Romero NB, Mezmezian M, Fidzianska A. Main steps of skeletal muscle development in the human: morphological analysis and ultrastructural characteristics of developing human muscle. Handb Clin Neurol. 2013; 113: 1299–1310. [CrossRef] [Google scholar] [PubMed]
- Brennan KA, Olson DM, Symonds ME. Maternal nutrient restriction alters renal development and blood pressure regulation of the offspring. Proc Nutr Soc. 2006; 65: 116–124. [CrossRef] [Google scholar] [PubMed]
- Venuti JM, Morris JH, Vivian JL, Olson EN, Klein WH. Myogenin is required for late but not early aspects of myogenesis during mouse development. J Cell Biol. 1995; 128: 563–576. [CrossRef] [Google scholar] [PubMed]
- Symonds ME, Stephenson T, Gardner DS, Budge H. Long-term effects of nutritional programming of the embryo and fetus: mechanisms and critical windows. Reprod Fertil Dev. 2006; 19: 53–63. [CrossRef] [Google scholar] [PubMed]
- Bryden M, Evans HE, Binns W. Embryology of the sheep. I. Extraembryonic membranes and the development of body form. J Morphol. 1972; 138: 169–185. [CrossRef] [Google scholar] [PubMed]
- Bryden M, Evans HE, Binns W. Embryology of the sheep. II. The alimentary tract and associated glands. J Morphol. 1972; 138: 187–205. [CrossRef] [Google scholar] [PubMed]
- Pinkerton KE, Joad JP. The mammalian respiratory system and critical windows of exposure for children’s health. Environ Health Perspect. 2000; 108: 457. [Google scholar]
- Howdeshell KL. A model of the development of the brain as a construct of the thyroid system. Environ Health Perspect. 2002; 110: 337. [CrossRef] [Google scholar] [PubMed]
- Nagy A. Manipulating the mouse embryo: a laboratory manual. New York: Cold Spring Harbor Laboratory Press; 2003. [Google scholar]
- Theiler K, others. The house mouse: Development and normal stages from fertilization to 4 weeks of age. New York: Springer-Verlag; 1972. [Google scholar]
- Wu G, Bazer F, Wallace J, Spencer T. Board-invited review: Intrauterine growth retardation: Implications for the animal sciences. J Anim Sci. 2006; 84: 2316–2337. [CrossRef] [Google scholar] [PubMed]
- Faraci M, Renda E, Monte S, Di Prima FA, Valenti O, De Domenico R, et al. Fetal growth restriction: current perspectives. J Prenat Med. 2011; 5: 31. [Google scholar]
- Satterfield MC, Bazer FW, Spencer TE, Wu G. Sildenafil citrate treatment enhances amino acid availability in the conceptus and fetal growth in an ovine model of intrauterine growth restriction. J Nutr. 2010; 140: 251–258. [CrossRef] [Google scholar] [PubMed]
- Lassala A, Bazer FW, Cudd TA, Datta S, Keisler DH, Satterfield MC, et al. Parenteral administration of L-arginine prevents fetal growth restriction in undernourished ewes. J Nutr. 2010; 140: 1242–1248. [CrossRef] [Google scholar] [PubMed]
- Luther J, Milne J, Aitken R, Matsuzaki M, Reynolds L, Redmer D, et al. Placental growth, angiogenic gene expression, and vascular development in undernourished adolescent sheep. Biol Reprod. 2007; 77: 351–357. [CrossRef] [Google scholar] [PubMed]
- Kelly RW. Nutrition and placental development. Proc Nutr Soc Aust. 1992; 17: 203–211. [Google scholar]
- Chandler K, Leury B, Bird A, Bell A. Effects of undernutrition and exercise during late pregnancy on uterine, fetal and uteroplacental metabolism in the ewe. Br J Nutr. 1985; 53: 625–635. [CrossRef] [Google scholar] [PubMed]
- Kwon H, Ford SP, Bazer FW, Spencer TE, Nathanielsz PW, Nijland MJ, et al. Maternal nutrient restriction reduces concentrations of amino acids and polyamines in ovine maternal and fetal plasma and fetal fluids. Biol Reprod. 2004; 71: 901–908. [CrossRef] [Google scholar] [PubMed]
- Zhu MJ, Ford SP, Means WJ, Hess BW, Nathanielsz PW, Du M. Maternal nutrient restriction affects properties of skeletal muscle in offspring. J Physiol. 2006; 575: 241–250. [CrossRef] [Google scholar] [PubMed]
- Fahey A, Brameld J, Parr T, Buttery P. The effect of maternal undernutrition before muscle differentiation on the muscle fiber development of the newborn lamb. J Anim Sci. 2005; 83: 2564–2571. [CrossRef] [Google scholar] [PubMed]
- Zhu MJ, Ford SP, Nathanielsz PW, Du M. Effect of maternal nutrient restriction in sheep on the development of fetal skeletal muscle. Biol Reprod. 2004; 71: 1968–1973. [CrossRef] [Google scholar] [PubMed]
- George LA, Zhang L, Tuersunjiang N, Ma Y, Long NM, Uthlaut AB, et al. Early maternal undernutrition programs increased feed intake, altered glucose metabolism and insulin secretion, and liver function in aged female offspring. Am J Physiol Regul Integr Comp Physiol. 2012; 302: R795–R804. [CrossRef] [Google scholar] [PubMed]
- Gardner DS, Tingey K, Van Bon BWM, Ozanne SE, Wilson V, Dandrea J, et al. Programming of glucose-insulin metabolism in adult sheep after maternal undernutrition. Am J Physiol Regul Integr Comp Physiol. 2005; 289: R947–R954. [CrossRef] [Google scholar] [PubMed]
- Satterfield MC, Dunlap KA, Keisler DH, Bazer FW, Wu G. Arginine nutrition and fetal brown adipose tissue development in nutrient-restricted sheep. Amino Acids. 2013; 45: 489–499. [CrossRef] [Google scholar] [PubMed]
- Vonnahme K, Luther J, Reynolds L, Hammer C, Carlson D, Redmer D, et al. Impacts of maternal selenium and nutritional level on growth, adiposity, and glucose tolerance in female offspring in sheep. Domest Anim Endocrinol. 2010; 39: 240–248. [CrossRef] [Google scholar] [PubMed]
- Osgerby JC, Wathes DC, Howard D, Gadd TS. The effect of maternal undernutrition on ovine fetal growth. J Endocrinol. 2002; 173: 131–141. [CrossRef] [Google scholar] [PubMed]
- Vonnahme KA. Maternal undernutrition from early- to mid-gestation leads to growth retardation, cardiac ventricular hypertrophy, and increased liver weight in the fetal sheep. Biol Reprod. 2003; 69: 133–140. [CrossRef] [Google scholar] [PubMed]
- Hyatt M, Gopalakrishnan G, Bispham J, Gentili S, McMillen IC, Rhind S, et al. Maternal nutrient restriction in early pregnancy programs hepatic mRNA expression of growth-related genes and liver size in adult male sheep. J Endocrinol 2007; 192: 87–97. [CrossRef] [Google scholar] [PubMed]
- Hyatt M, Gardner D, Sebert S, Wilson V, Davidson N, Nigmatullina Y, et al. Suboptimal maternal nutrition, during early fetal liver development, promotes lipid accumulation in the liver of obese offspring. Reproduction. 2011; 141: 119–126. [CrossRef] [Google scholar] [PubMed]
- Chadio S, Kotsampasi B, Taka S, Liandris E, Papadopoulos N, Plakokefalos E. Epigenetic changes of hepatic glucocorticoid receptor in sheep male offspring undernourished in utero. Reprod Fertil Dev. 2017; 29: 1995–2004. [CrossRef] [Google scholar] [PubMed]
- Darby JRT, McMillen IC, Morrison JL. Maternal undernutrition in late gestation increases IGF2 signalling molecules and collagen deposition in the right ventricle of the fetal sheep heart. J Physiol. 2018; 596: 2345–2358. [CrossRef] [Google scholar] [PubMed]
- Shukla P, Ghatta S, Dubey N, Lemley CO, Johnson ML, Modgil A, et al. Maternal nutrient restriction during pregnancy impairs an endothelium-derived hyperpolarizing factor-like pathway in sheep fetal coronary arteries. Am J Physiol Heart Circ Physiol. 2014; 307: H134–H142. [CrossRef] [Google scholar] [PubMed]
- Gilbert JS, Lang AL, Grant AR, Nijland MJ. Maternal nutrient restriction in sheep: hypertension and decreased nephron number in offspring at 9 months of age. J Physiol. 2005; 565: 137–147. [CrossRef] [Google scholar] [PubMed]
- Dunford LJ, Sinclair KD, Kwong WY, Sturrock C, Clifford BL, Giles TC, et al. Maternal protein-energy malnutrition during early pregnancy in sheep impacts the fetal ornithine cycle to reduce fetal kidney microvascular development. FASEB J. 2014; 28: 4880–4892. [CrossRef] [Google scholar] [PubMed]
- Lloyd LJ, Foster T, Rhodes P, Rhind SM, Gardner DS. Protein-energy malnutrition during early gestation in sheep blunts fetal renal vascular and nephron development and compromises adult renal function. J Physiol. 2012; 590: 377–393. [CrossRef] [Google scholar] [PubMed]
- Ford S, Hess B, Schwope M, Nijland M, Gilbert J, Vonnahme K, et al. Maternal undernutrition during early to mid-gestation in the ewe results in altered growth, adiposity, and glucose tolerance in male offspring. J Anim Sci. 2007; 85: 1285–1294. [CrossRef] [Google scholar] [PubMed]
- Adam C, Williams P, Milne J, Aitken R, Wallace J. Orexigenic gene expression in late gestation ovine foetal hypothalamus is sensitive to maternal undernutrition and realimentation. J Neuroendocrinol. 2015; 27: 765–771. [CrossRef] [Google scholar] [PubMed]
- Gluckman PD, Hanson MA, Spencer HG. Predictive adaptive responses and human evolution. Trends Ecol Evol. 2005; 20: 527–533. [CrossRef] [Google scholar] [PubMed]
- Gluckman PD, Hanson MA, Beedle AS. Early life events and their consequences for later disease: A life history and evolutionary perspective. Am J Hum Biol. 2007; 19: 1–19. [CrossRef] [Google scholar] [PubMed]
- Gluckman PD, Hanson MA. Developmental origins of disease paradigm: A mechanistic and evolutionary perspective. Pediatr Res. 2004; 56: 311. [CrossRef] [Google scholar] [PubMed]
- Dunlap KA, Brown JD, Keith AB, Satterfield MC. Factors controlling nutrient availability to the developing fetus in ruminants. J Anim Sci Biotechnol. 2015; 6: 16. [CrossRef] [Google scholar] [PubMed]
- Sosnowski DW, Booth C, York TP, Amstadter AB, Kliewer W. Maternal prenatal stress and infant DNA methylation: A systematic review. Dev Psychobiol. 2018; 60: 127–139. [CrossRef] [Google scholar] [PubMed]
- Mazzio EA, Soliman KF. Basic concepts of epigenetics: Impact of environmental signals on gene expression. Epigenetics. 2012; 7: 119–130. [CrossRef] [Google scholar] [PubMed]
- Yamada L, Chong S. Epigenetic studies in developmental origins of health and disease: Pitfalls and key considerations for study design and interpretation. J Dev Orig Health Dis. 2017; 8: 30–43. [CrossRef] [Google scholar] [PubMed]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet. 2003; 33: 245. [CrossRef] [Google scholar] [PubMed]
- Ji Y, Wu Z, Dai Z, Sun K, Wang J, Wu G. Nutritional epigenetics with a focus on amino acids: Implications for the development and treatment of metabolic syndrome. J Nutr Biochem. 2016; 27: 1–8. [CrossRef] [Google scholar] [PubMed]
- Guéant JL, Namour F, Guéant-Rodriguez RM, Daval JL. Folate and fetal programming: A play in epigenomics?. Trends Endocrinol Metab. 2013; 24: 279–289. [CrossRef] [Google scholar] [PubMed]
- Sinclair KD, Allegrucci C, Singh R, Gardner DS, Sebastian S, Bispham J, et al. DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc Natl Acad Sci USA. 2007; 104: 19351–19356. [CrossRef] [Google scholar] [PubMed]
- Dong X, Weng Z. The correlation between histone modifications and gene expression. Epigenomics. 2013; 5: 113–116. [CrossRef] [Google scholar] [PubMed]
- Vickers MH. Early life nutrition, epigenetics and programming of later life disease. Nutrients. 2014; 6: 2165–2178. [CrossRef] [Google scholar] [PubMed]
- Molina-Serrano D, Schiza V, Kirmizis A. Cross-talk among epigenetic modifications: Lessons from histone arginine methylation. Biochem Soc Trans. 2013; 41: 751–759 [CrossRef] [Google scholar] [PubMed]
- Makowski EL, Meschia G, Droegemueller W, Battaglia FC. Measurement of umbilical arterial blood flow to the sheep placenta and fetus in utero. Obstet Gynecol Surv. 1969; 24: 602–607. [CrossRef] [Google scholar]
- Borowicz PP, Arnold DR, Johnson ML, Grazul-Bilska AT, Redmer DA, Reynolds LP. Placental growth throughout the last two thirds of pregnancy in sheep: Vascular development and angiogenic factor expression. Biol Reprod. 2007; 76: 259–267. [CrossRef] [Google scholar] [PubMed]
- Lang U, Baker RS, Braems G, Zygmunt M, Künzel W, Clark KE. Uterine blood flow-a determinant of fetal growth. Eur J Obstet Gynecol Reprod Biol. 2003; 110: S55–S61. [CrossRef] [Google scholar] [PubMed]
- Hawkes N. Trial of Viagra for fetal growth restriction is halted after baby deaths. BMJ. 2018; 362: k3247. [CrossRef] [Google scholar] [PubMed]
- Regnault TRH, Friedman JE, Wilkening RB, Anthony RV, Hay WW. Fetoplacental transport and utilization of amino acids in IUGR-a review. Placenta. 2005; 26: S52–S62. [CrossRef] [Google scholar] [PubMed]
- Paolini CL, Meschia G, Fennessey PV, Pike AW, Teng C, Battaglia FC, et al. An in vivo study of ovine placental transport of essential amino acids. Am J Physiol Endocrinol Metab. 2001; 280: E31–E39. [CrossRef] [Google scholar] [PubMed]
- Brown LD. Endocrine regulation of fetal skeletal muscle growth: Impact on future metabolic health. J Endocrinol. 2014; 221: R13–R29. [Google scholar]
- Du M, Tong J, Zhao J, Underwood K, Zhu M, Ford S, et al. Fetal programming of skeletal muscle development in ruminant animals. J Anim Sci. 2010; 88: E51–E60. [CrossRef] [Google scholar] [PubMed]
- Frontera WR, Ochala J. Skeletal muscle: A brief review of structure and function. Calcif Tissue Int. 2015; 96: 183–195. [CrossRef] [Google scholar] [PubMed]
- Wu G. Functional amino acids in nutrition and health. Amino Acids. 2013; 45: 407–411. [CrossRef] [Google scholar] [PubMed]
- Du M, Zhu M, Means W, Hess B, Ford S. Nutrient restriction differentially modulates the mammalian target of rapamycin signaling and the ubiquitin-proteasome system in skeletal muscle of cows and their fetuses. J Anim Sci. 2005; 83: 117–123. [CrossRef] [Google scholar] [PubMed]
- Fisher G, Windham ST, Griffin P, Warren JL, Gower BA, Hunter GR. Associations of human skeletal muscle fiber type and insulin sensitivity, blood lipids, and vascular hemodynamics in a cohort of premenopausal women. Eur J Appl Physiol. 2017; 117: 1413–1422. [CrossRef] [Google scholar] [PubMed]
- Cassuto H, Kochan K, Chakravarty K, Cohen H, Blum B, Olswang Y, et al. Glucocorticoids regulate transcription of the gene for phosphoenolpyruvate carboxykinase (GTP) in the liver via an extended glucocorticoid regulatory unit. J Biol Chem. 2005; 280: 33873–33884. [CrossRef] [Google scholar] [PubMed]
- Barker DJ, Godfrey KM, Gluckman PD, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. The Lancet. 1993; 341: 938–941. [CrossRef] [Google scholar] [PubMed]
- Dong F, Ford SP, Fang CX, Nijland MJ, Nathanielsz PW, Ren J. Maternal nutrient restriction during early to mid gestation up-regulates cardiac insulin-like growth factor (IGF) receptors associated with enlarged ventricular size in fetal sheep. Growth Horm IGF Res. 2005; 15: 291–299. [CrossRef] [Google scholar] [PubMed]
- Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure: Less of one, more the other? Am J Hypertens. 1988; 1: 335–347. [CrossRef] [Google scholar] [PubMed]
- Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004; 84: 277–359. [CrossRef] [Google scholar] [PubMed]
- Satterfield MC, Wu G. Brown adipose tissue growth and development: Significance and nutritional regulation. Front Biosci (Landmark Ed). 2011; 16: 1589–1608. [CrossRef] [Google scholar] [PubMed]
- Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009; 58: 1526–1531. [CrossRef] [Google scholar] [PubMed]
- Yoneshiro T, Aita S, Matsushita M, Kameya T, Nakada K, Kawai Y, et al. Brown adipose tissue, whole-body energy expenditure, and thermogenesis in healthy adult men. Obesity. 2011; 19: 13–16. [CrossRef] [Google scholar] [PubMed]
- Cypess AM, Kahn CR. Brown fat as a therapy for obesity and diabetes. Curr Opin Endocrinol Diabetes Obes. 2010; 17: 143. [CrossRef] [Google scholar] [PubMed]
- Vijgen GH, Bouvy ND, Teule GJ, Brans B, Schrauwen P, van Marken Lichtenbelt WD. Brown adipose tissue in morbidly obese subjects. PLoS One. 2011; 6: e17247. [CrossRef] [Google scholar] [PubMed]
- Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, et al. Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest. 2013; 123: 3404–3408. [CrossRef] [Google scholar] [PubMed]
- Satterfield MC, Dunlap KA, Keisler DH, Bazer FW, Wu G. Arginine nutrition and fetal brown adipose tissue development in diet-induced obese sheep. Amino Acids. 2012; 43: 1593–1603. [CrossRef] [Google scholar] [PubMed]
- Ma X, Han M, Li D, Hu S, Gilbreath KR, Bazer FW, et al. l-Arginine promotes protein synthesis and cell growth in brown adipocyte precursor cells via the mTOR signal pathway. Amino Acids. 2017; 49: 957–964. [CrossRef] [Google scholar] [PubMed]
- Henry BA, Pope M, Birtwistle M, Loughnan R, Alagal R, Fuller-Jackson J-P, et al. Ontogeny and thermogenic role for sternal fat in female sheep. Endocrinology. 2017; 158: 2212–2225. [CrossRef] [Google scholar] [PubMed]
- Attig L, Gabory A, Junien C. Early nutrition and epigenetic programming: Chasing shadows. Curr Opin Clin Nutr Metab Care. 2010; 13: 284–293. [CrossRef] [Google scholar] [PubMed]

