

In Silico Detection and FISH Analysis to Determine Location of miRNAs in Solea senegalensis Chromosomes Using BACs
Alberto Arias-Pérez 1, *![]() , Daniel Ramírez-Torres 1
, Daniel Ramírez-Torres 1![]() , María E. Rodríguez 1
, María E. Rodríguez 1![]() , Silvia Portela-Bens 1
, Silvia Portela-Bens 1![]() , Emilio García-Suarez 1
, Emilio García-Suarez 1![]() , Manuel A. Merlo 1
, Manuel A. Merlo 1![]() , Aglaya García-Angulo 1
, Aglaya García-Angulo 1![]() , Ismael Cross 1
, Ismael Cross 1![]() , Thomas Liehr 2
, Thomas Liehr 2![]() , Laureana Rebordinos 1
, Laureana Rebordinos 1![]()
- Area de Genética, Departamento de Biomedicina y Biotecnología, INMAR, UCA, Puerto Real, Cádiz, Spain
- Jena University Hospital, Friedrich Schiller University, Institute of Human Genetics, Jena, Germany
* Correspondence: Alberto Arias-Pérez![]()
Received: July 31, 2018 | Accepted: September 27, 2018 | Published: October 24, 2018
OBM Genetics 2018, Volume 2, Issue 4 doi:10.21926/obm.genet.1804044
Academic Editors: Thomas Liehr
Special Issue: Applications of Fluorescence in Situ Hybridization
Recommended citation: Arias-Pérez A, Ramírez-Torres D, E. Rodríguez M, Portela-Bens S, García-Suarez E, Merlo MA, García-Angulo A, Cross I, Liehr T, Rebordinos L. In Silico Detection and FISH Analysis to Determine Location of miRNAs in Solea senegalensis Chromosomes Using BACs. OBM Genetics 2018;2(4):044; doi:10.21926/obm.genet.1804044.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Background: MicroRNAs (miRNAs) are small, non-coding RNAs that play a very important role in gene expression by regulating mRNA cleavage and translation. The Senegalese sole, Solea senegalensis (Kaup 1858), is a flatfish species that shows great potential for marine aquaculture. Nevertheless, the existence of sexual dysfunction of males reared in captivity, high larval mortality, and diseases have hampered its production. The integration of sequence information with data on chromosomal physical location is useful for understanding genetic processes and is essential for comparative analysis, quantitative trait locus (QTL) mapping, and positional cloning of genes. Methods: In this study we employed an in silico approach to identify potential miRNAs in an S. senegalensis BAC library. Mature sequences were obtained from miRBase and other sources, and a BLAST analysis was carried out against sequences from an S. senegalensis BAC library. Sequences showing similarity were extracted and subjected to analysis of their secondary structure. This information was combined with BAC-FISH data to assign miRNAs to particular chromosomes. In addition, a phylogenetic analysis of some miRNA families was carried out and target genes were predicted against a reference transcriptome. Results: After secondary structure analysis, 45 groups of sequences showed similarity to mature miRNAs. Of these, 19 were assigned to Senegalese Sole chromosomes using BAC-FISH data, and 34 unique mature sequences targeted 12387 sequences from an S. senegalensis reference transcriptome. Phylogenetic trees showed different levels of support for clustering among types of miRNAs; S. senegalensis sequences were usually close to those of other Pleuronectiformes. Conclusions: These results will be helpful for understanding how miRNAs affect gene expression in this species and the biological processes involved in its aquaculture. Nevertheless, experimental validation using RT-PCR, northern blotting, or microarrays, is required to confirm the predictions made here.
Keywords
MicroRNAs; BAC-FISH; solea senegalensis; non-coding RNA; flat fish
1. Introduction
MicroRNAs (miRNAs) are small, non-coding RNAs with lengths ranging from 18 to 25 nucleotides (nt). These RNAs play a very important role in gene expression by regulating mRNA cleavage and translation. The biogenesis of miRNA is a complex process and several pathways are known [1,2]. As a general rule, miRNA synthesis begins in the nucleus where pri-miRNAs (capped and polyadenylated primary nascent transcripts) are transcribed by RNA polymerase II. Then, these pri-miRNAs are processed to shorter precursor molecules (approximately 70 nucleotides) known as pre-miRNAs. This step is carried out by Drosha, an RNAse type III enzyme, along with several other polypeptides. Later the pre-miRNA is transported to the cytoplasm where Dicer and associated proteins generate a duplex that, on average, is 22 nt long. Usually one of the strands will form part of an active miRNA-induced silencing complex (miRISC) [1,2,3].
MicroRNAs mainly influence gene expression by post-transcriptional repression of mRNA in cytoplasm, but some of them are imported back into the nucleus where they regulate the expression of non-coding RNAs [4]. The regulation of mRNA cleavage and translation is related to base complementarity between the miRNA and its target. A perfect pairing induces target degradation while imperfect pairing inhibits translation elongation [5]. Nucleotides at positions 2-8 from the 5’ end of a mature miRNA play a fundamental role in miRNA-to-target mRNA pairing. This “seed” sequence is usually highly conserved among taxa, and is used to classify miRNAs into families [6].
These non-coding RNAs were initially discovered in Caenorhabditis elegans [7,8] and it took some years to uncover the importance that these non-coding RNAs have in gene expression. Currently, there is extensive ongoing research in both animals and plants in relation to the role of miRNAs in different biological processes including skeletal muscle development [9], cancer [10], and responses to stress [11]. In teleosts, the number of studies is still quite limited (but see [2], [12], and [13]). Currently, the number of mature miRNA sequences available at miRBase (v. 22) is 3687, corresponding to only 16 species. It is clear that considerable effort is needed to comprehend how miRNAs generally affect gene expression in fish.
The Senegalese sole, Solea senegalensis (Kaup 1858), is a flatfish species belonging to the Pleuronectiformes order. It is distributed in the Atlantic Ocean from the Gulf of Biscay to the Northwest coast of Africa, and it is less frequently found in the Western Mediterranean [14]. It is a predominantly littoral and benthonic marine species that shows great potential for marine aquaculture diversification due to its growth rates and flesh quality [15]. Nevertheless, the existence of sexual dysfunction of males reared in captivity, high larval mortality, and diseases have hampered its production [16]. In recent years, an increasing number of studies have assessed different genetic and genomic aspects of S. senegalensis such as transcriptomics [17], integration of genetic maps [18], and cytogenomics [19]. There are also various studies assessing gene expression [20], but to date only one study has explored miRNA sequences in S. senegalensis [21]. This study assessed the role of miRNAs in the thermal plasticity of growth, identifying 320 conserved miRNAs and observed a strong influence of temperature on expression of miRNAs.
The integration of sequence information with data on chromosomal physical location is useful for understanding genetic processes and is essential for comparative analysis, QTL mapping, and positional cloning of genes [22]. The combination of fluorescence in situ hybridization (FISH) and bacterial artificial chromosome (BAC) libraries provides an efficient approach for anchoring genomic and linkage sequence data onto physical chromosomes [23].
In this study, we employed an in silico approach to identify potential miRNAs and their target genes in an S. senegalensis BAC library. This information was then combined with BAC-FISH data. The results will help improve the understanding of the regulatory roles of these miRNAs in Senegalese Sole and other fish, but will also be useful for understanding their evolution and distribution.
2. Materials and Methods
2.1. In Silico Detection of Potential miRNAs and FISH Analysis to Determine Location
Mature miRNA sequences (35828 entries in 223 species) were downloaded from miRBase (Release 22, March 2018; http://www.mirbase.org). In addition, mature sequences from different fish species were also considered: Culter alburnus [24], Cynoglossus semilaevis [25], Cyprinus carpio [26], Hippoglossus hippoglossus [27], Paralichthys olivaceus [28,29], Scophthalmus maximus [30], and Solea senegalensis [21]. To avoid redundant results, non-unique mature miRNA sequences were merged (FASTA Unique sequences, https://www.ncbi.nlm.nih.gov/CBBresearch/Spouge/html_ncbi/html/fasta/uniqueseq.cgi).
A nucleotide database was created containing 2981 sequences from 91 BAC clones of a Solea senegalensis BAC library with construction details described in [31], using the basic local alignment search tool (NCBI suite BLAST 2.6.0+). This database was then interrogated using blastn command for potentially conserved miRNAs, employing the following parameters: task “blastn”, percent identity of 0.7, percent query coverage per high-scoring segment pair of 1, an expectation value threshold of 0.01, and low complexity sequence data was filtered using the DUST algorithm. All BLAST results with a maximum of 4 mismatches and no gaps were used for further analysis. Potential mature miRNAs and surrounding flanking regions (100 bp on each side, when possible) were extracted with the BLAST tool blastdbcmd. These sequences were used with MIReNA 2.0 [32], which filters secondary structures using five criteria to predict pre-miRNAs. The program RNAfold 2.4.6 [33] from the ViennaRNA package was used to generate RNA secondary structure plots. Finally, pre-miRNAs were manually inspected to remove possible false positives.
BAC-FISH data was obtained from previous works [18,23,31] and by employing the FISH techniques described in [31]. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
2.2. Phylogenetic Analysis of miRNAs
To study the evolutionary relationship among the potential miRNAs detected for S. senegalensis and those of other teleosts, the stem-loop sequences available at miRBase were downloaded. When possible, stem-loop sequences from the references mentioned in the previous section were also used. The MEGA X software [34] was used to align the sequences (via the MUSCLE algorithm with default parameters) and to build a tree by the maximum likelihood method (by means of partial deletion with site coverage cut-off of 95% with 1000 bootstrap replications).
2.3. Target Prediction
The reference transcriptome for S. senegalensis (v4.1) was downloaded from Solea DB [17] and used for target prediction using miRanda v3.3a [35]. Only the last part of the sequences was used, given that the 3'-UTR region for most S. senegalensis genes has not yet been resolved as well as to speed up the computational analysis. All sequences were conservatively trimmed to a maximum of 1000 nt, given that previous data on teleosts showed a median 3'-UTR length of 795 nt [36]. The parameters employed were: minimum score of 50, free energy ≤ 20 kcal/mol, and strict alignment in the seed region.
3. Results and Discussion
3.1. In Silico Detection of Potential miRNAs
The advent of NGS technologies and the feasibility of large-scale genomic projects have made sequencing data available on an unprecedented scale. This makes it possible to search different types of sequences and even to analyze the chromosome structure of non-model organisms. In the present study, an in silico approach was used to screen miRNAs, which play a fundamental role in the regulation of gene expression, in BAC data of S. senegalensis. The BLAST analysis of 2981 contigs (with an average length of about 4800 bp) from 91 clones of a BAC library of S. senegalensis yielded 272 matches. These matches, together with some flanking sequences, were extracted for pre-miRNA and secondary structure prediction with MIReNA and RNAfold. This resulted in 124 potential miRNAs; however, given that some of them map to approximately the same position due to differences in the mature sequence length, 45 groups were obtained (Table 1 and Table S1). The largest group contained 16 mature sequences ranging in length from 20 to 23 bp. When considering only one mature sequence per group, that being the one with the lowest e-value, most of them were observed once but others were found several times: for example, miR-467g and miR-8915 appeared nine and five times, respectively. Other miRNAs appeared three times (miR-669l-3p, miR-467e-3p) and twice (let-7f, dme-miR-4968-3p, cse-PC-5722-5p, and let-7f-5p).
Table 1 Potential miRNAs identified in Solea senegalensis BAC sequences (full details in Table S1). Group numbers (column 1) with two integers separated by a dot indicate multiple mature miRNAs matching to the same or almost the same position. When a miRNA name (column 3) corresponds to a group name (column 2), this means that this sequence (column 4) is the 5’ or 3’ arm of the other mature sequence. Sequence names (column 4) contain the following four items of information separated by dashes: chromosome number (99 indicates hybridization to multiple chromosomes, na indicates no hybridization); BAC name (plate number is the first two characters and plate position is the last three characters); sequencing batch; and contig number. Other keys given at foot of table.
| Group | Name | miRNA homolog | Sequence | Positions | Side | NM | Strand | LP (nt) | A+U | MFE | MFEI |
| 1 | sse-miR-8915-1 | efu-miR-8915 | 01-05K05-02-317 | 231-253 | 3' | 2 | plus | 80 | 0.33 | -38.9 | 0.73 |
| 2 | sse-miR-8915-2 | efu-miR-8915 | 01-12D22-02-001 | 8284-8306 | 3' | 2 | minus | 81 | 0.32 | -40.2 | 0.73 |
| 3 | sse-miR-8915-3 | efu-miR-8915 | 01-12D22-02-001 | 16449-16471 | 3' | 2 | minus | 81 | 0.32 | -40.2 | 0.73 |
| 4 | sse-miR-8915-4 | efu-miR-8915 | 01-12D22-02-002 | 1871-1893 | 3' | 2 | minus | 81 | 0.33 | -40.2 | 0.74 |
| 5 | sse-miR-8915-5 | efu-miR-8915 | 01-12D22-02-002 | 8073-8095 | 3' | 2 | plus | 81 | 0.32 | -40.2 | 0.73 |
| 6.1 | sse-miR-460 | cca-miR-460-5p | 01-48P07-07-005 | 809-830 | 5' | 0 | plus | 60 | 0.55 | -22.5 | 0.83 |
| 7.1 | As 6.1 | sma-miR-460-3p* | 01-48P07-07-005 | 844-865 | 3' | 0 | plus | 60 | 0.55 | -22.5 | 0.83 |
| 8 | sse-PC-5722-1 | cse-PC-5722-5p | 01-73B07-09-003 | 1585-1602 | 5' | 0 | plus | 112 | 0.16 | -69.3 | 0.74 |
| 9 | sse-miR-9388 | dme-miR-9388-5p | 04-08A23-03-002 | 10919-10941 | 3' | 2 | minus | 60 | 0.75 | -23.3 | 1.55 |
| 10.1 | sse-miR-153b | hhi-miR-153b | 04-12D24-04-001 | 93475-93497 | 3' | 0 | minus | 91 | 0.58 | -39.5 | 1.03 |
| 11.1 | As 10.1 | dre-miR-153b-5p | 04-12D24-04-001 | 93514-93535 | 5' | 0 | minus | 91 | 0.58 | -39.5 | 1.03 |
| 12 | sse-PC-2293 | cse-PC-2293-5p | 06-10K23-06-002 | 20514-20535 | 5' | 0 | plus | 80 | 0.56 | -36.1 | 1.03 |
| 13.1 | sse-PC-15046 | cse-PC-15046-3p | 06-10K23-06-002 | 20555-20576 | 3' | 0 | plus | 80 | 0.56 | -36.1 | 1.03 |
| 14 | sse-miR-4968 | dme-miR-4968-3p | 12-35D17-07-001 | 37843-37864 | 3' | 1 | plus | 111 | 0.41 | -45.9 | 0.70 |
| 15.1 | sse-miR-467g-1 | mmu-miR-467g | 16-09N08-05-002 | 8062-8082 | 5' | 0 | plus | 86 | 0.73 | -36.8 | 1.58 |
| 16 | sse-miR-466f | mmu-miR-466f | 18-36M02-06-001 | 22980-23001 | 5' | 1 | plus | 125 | 0.53 | -41 | 0.70 |
| 17.1 | sse-miR-467g-2 | mmu-miR-467g | 19-12K06-05-001 | 6816-6836 | 5' | 0 | plus | 86 | 0.73 | -36.8 | 1.58 |
| 18 | sse-miR-467g-3 | mmu-miR-467g | 21-63A03-05-013 | 1740-1760 | 5' | 1 | minus | 64 | 0.81 | -24.2 | 1.99 |
| 19 | sse-miR-5p-163498.9 | pol-miR-5p-163498.9 | 99-08O07-04-004 | 3651-3671 | 5' | 1 | minus | 61 | 0.62 | -22.4 | 0.97 |
| 20 | sse-miR-430b | hhi-miR-430b | na-09B02-02-015 | 217-238 | 3' | 0 | plus | 78 | 0.55 | -28.3 | 0.81 |
| 21.1 | sse-miR-467g-4 |
mmu-miR-467g |
na-12K16-02-001 | 9843-9863 | 5' | 0 | minus | 86 | 0.73 | -36.8 | 1.58 |
| 22 | sse-PC-5722-2 | cse-PC-5722-5p | na-31B01-07-001 | 6483-6500 | 5' | 0 | plus | 112 | 0.16 | -69.3 | 0.74 |
| 23 | sse-miR-467g-5 | mmu-miR-467g | na-42P04-09-004 | 1738-1758 | 3' | 1 | plus | 60 | 0.8 | -27.6 | 2.30 |
| 24 | sse-miR-3584 | rno-miR-3584-5p | na-46K16-09-022 | 3166-3183 | 3' | 0 | plus | 69 | 0.35 | -34 | 0.76 |
| 25.1 | sse-let-7e | aca-let-7e-5p | na-50K03-08-014 | 656-677 | 5' | 0 | plus | 71 | 0.59 | -27.2 | 0.93 |
| 26.1 | As 25.1 | aca-let-7e-3p | na-50K03-08-014 | 704-727 | 3' | 1 | plus | 72 | 0.58 | -27.2 | 0.90 |
| 27.1 | sse-let-7a | prd-let-7-5p | na-50K03-08-014 | 889-911 | 5' | 0 | plus | 85 | 0.58 | -40.9 | 1.15 |
| 28.1 | As 27.1 | pol-let-7a-3-p3 | na-50K03-08-014 | 944-965 | 3' | 0 | plus | 85 | 0.58 | -40.9 | 1.15 |
| 29.1 | sse-miR-205 | efu-miR-205 | na-53B20-09-006 | 524-546 | 5' | 0 | plus | 88 | 0.53 | -35.7 | 0.86 |
| 30.1 | As 29.1 | ola-miR-205 | na-53B20-09-006 | 561-582 | 3' | 0 | plus | 88 | 0.53 | -35.7 | 0.86 |
| 31 | sse-miR-467g-6 | mmu-miR-467g | na-53B20-09-019 | 7147-7167 | 5' | 1 | minus | 94 | 0.84 | -32 | 2.13 |
| 32 | sse-miR-1285 | cgr-miR-1285 | na-53I12-09-092 | 358-381 | 3' | 2 | plus | 102 | 0.42 | -47.9 | 0.81 |
| 33 | sse-miR-467g-7 | mmu-miR-467g | na-59B23-09-003 | 12950-12970 | 3' | 1 | minus | 94 | 0.85 | -46.3 | 3.28 |
| 34 | sse-miR-467g-8 | mmu-miR-467g | na-65I16-09-001 | 2864-2884 | 5' | 1 | minus | 86 | 0.9 | -31.8 | 3.70 |
| 35 | sse-miR-466b-2 | rno-miR-466b-2-3p | na-68G04-09-010 | 2229-2249 | 3' | 1 | minus | 113 | 0.82 | -45.1 | 2.22 |
| 36 | sse-miR-467g-9 | mmu-miR-467g | na-68G04-09-010 | 2402-2422 | 5' | 1 | minus | 105 | 0.81 | -37.4 | 1.87 |
| 37 | sse-miR-4968 | dme-miR-4968-3p | na-74P22-09-003 | 1140-1161 | 5' | 1 | minus | 77 | 0.39 | -35.4 | 0.75 |
| 38.1 | sse-miR-454 | cse-miR-454-3p | na-74P22-09-005 | 612-636 | 3' | 0 | minus | 93 | 0.51 | -44.6 | 0.98 |
| 39.1 | As 38.1 | tgu-miR-454-5p | na-74P22-09-005 | 656-678 | 5' | 1 | minus | 93 | 0.51 | -44.6 | 0.98 |
| 40 | As 41.1 | ola-miR-301a-3p | na-74P22-09-005 | 6812-6833 | 3' | 1 | minus | 88 | 0.53 | -37.4 | 0.90 |
| 41.1 | sse-miR-301b | cal-miR-301b-5p-1 | na-74P22-09-005 | 6852-6873 | 5' | 0 | minus | 88 | 0.53 | -37.4 | 0.90 |
| 42.1 | sse-miR-130c | aca-miR-130c | na-74P22-09-005 | 7164-7185 | 3' | 0 | minus | 60 | 0.62 | -23 | 1.01 |
| 43.1 | As 42.1 | cal-miR-130a-5p-1 | na-74P22-09-005 | 7201-7222 | 5' | 0 | minus | 60 | 0.62 | -23 | 1.01 |
| 44.1 | sse-miR-130a | cal-miR-130a-5p-2 | na-74P22-09-006 | 606-627 | 5' | 0 | plus | 60 | 0.5 | -24.3 | 0.81 |
| 45.1 | As 44.1 | aca-miR-130b-3p | na-74P22-09-006 | 645-664 | 3' | 0 | plus | 60 | 0.5 | -24.3 | 0.81 |
Key: NM: number of mismatches; LP: length of the precursor; A+U: A + U content; MFE: minimum folding free energy (kcal/mol); MFEI: minimal folding free energy index. Species abbreviations: Abu Astatotilapia burtoni, Cca Cyprinus carpio, Dre Danio rerio, Fru Fugu rubripes, Gmo Gadus morhua, Hhi Hippoglossus hippoglossus, Ipu Ictalurus punctatus, Mze Metriaclima zebra, Nbr Neolamprologus brichardi, Ola Oryzias latipes, Oni Oreochromis niloticus, Pny Pundamilia nyererei, Pol Paralichthys olivaceus, Ssa Salmo salar, Sse Solea senegalensis, Tni Tetraodon nigroviridis.
Among the 124 potential miRNAs, 52 have been not reported in fish (Table S1) and the remaining ones have been reported in 1 to 13 species (average 2.75). The species showing the greatest number of records were C. carpio (22), P. olivaceus (22), Danio rerio (20), and S. senegalensis (19); the sequences identified in the greatest number of species were aca-let-7a-5p (13), aca-let-7e-5p (11), cal-let-7-5p-13 (11), cal-miR-153-3p-1 (10), aca-let-7c-5p (10), and aca-miR-205a (10). To date, only one work has studied miRNAs in S. senegalensis [21], and the information available for the teleost species is limited (but see [3,12,13]). Therefore, the results obtained here will contribute to a better understanding of the evolution of miRNAs in teleosts.
To assign miRNAs to a particular chromosome, BAC-FISH data was employed [18,23,31]. The BAC library was screened for genes of interest and then positive clones were sequenced and annotated. Following this, FISH probes were prepared and hybridization was carried out (Figure 1). Most of the groups (26 of 45) could not be assigned to a particular chromosome, but 8, 3, and 2 groups were assigned to chromosomes 1, 4, and 6, respectively. For other chromosomes (12, 16, 18, 19, and 21) only one group was observed. Finally, one group of miRNAs corresponded to a BAC that hybridizes with multiple chromosomes. Although chromosome 1 seems to have a higher amount of miRNAs, the amount of data available does not allow significant differences to be inferred. This is the case in D. rerio, where miRNA genes have shown unequal distribution among chromosomes [37]. The analysis of the distribution of miRNAs in the Senegalese sole chromosomes might help in studying the biological implications of the genomic context of miRNA. In addition, the integration of QTL and miRNA could be used to investigate biological aspects such as disease susceptibility [13].
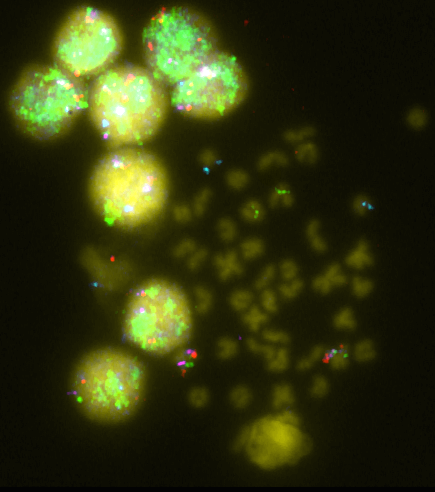
Figure 1 FISH of BACs 46P22 (green), 03C15 (pink), 08A23 (red) and 09J04 (blue).
The length of mature miRNAs ranged between 18 and 25 nt with a mode of 22; a length of 21 and 22 nt accounted for 52% of the 124 matches. The average length of the predicted precursor was 80 nt with a minimum and a maximum length of 60 and 125 nt, respectively. The secondary structures computed from these sequences showed the typical stem-loop miRNA structure (Supplementary data 1), except for sse-PC-5722-2 and sse-PC-5722-2 (groups 8 and 22) that had a more cruciform shape. This putative miRNA was described in C. semilaevis [25] by the sequencing of small RNA libraries. The precursor found in this study and in that of C. semilaevis differed markedly in size (112 vs 61 nt), and the alignment between these sequences was poor. This could be due to two distinct genomic loci expressing identical mature sequences or to the precursor found here belonging to another RNA class.
The placement of the mature miRNA sequences on the 5' or 3' arm of the stem-loop structure was almost equally distributed among the groups of miRNAs (23 and 22 results for the 3' and 5' arms, respectively), and the members of all groups were placed on the same side. Although usually only one of the strands of the miRNA duplex plays a role in the processes of gene regulation, there is evidence that some miRNAs can provide two mature functional miRNAs [38]. Our results revealed that there were several instances where significant matches for both strands of the precursor were observed: (a) 6 (cca-miR-460-5p) and 7 (sma-miR-460-3p*), (b) 10 (hhi-miR-153b) and 11 (dre-miR-153b-5p), (c) 12 (cse-PC-15046-3p) and 13 (cse-PC-2293-5p), (d) 25 (aca-let-7e-5p) and 26 (aca-let-7e-3p), (e) 27 (prd-let-7-5p) and 28 (pol-let-7a-3-p3), (f) 29 (efu-miR-205) and 30 (ola-miR-205), (g) 38 (cse-miR-454-3p) and 39 (tgu-miR-454-5p), (h) 40 (ola-miR-301a-3p) and 41 (cal-miR-301b-5p-1), and (i) 44 (cal-miR-130a-5p-2) and 45 (aca-miR-130b-3p).
Several miRNAs were located relatively close to one another: (a) sse-miR-8915-2 and sse-miR-8915-3; (b) sse-miR-8915-4 and sse-miR-8915-5; (c) sse-let-7e and sse-let-7a; and (d) sse-miR-454, sse-miR-301b, and sse-miR-130c. This suggests that they might belong to a miRNA gene cluster as has been observed for many miRNAs [3], which commonly form part of polycistronic transcripts. It is believed that these gene clusters could help coordinate the regulation of biological processes. The identification of the promotor regions of these miRNAs could help determine if these sequences form a true cluster.
In regard to the putative pre-miRNA, the minimum folding free energy for secondary structures ranged from -69.3 to -22.4 kcal/mol (with an average of -34.79), and the adenosine and uridine content (A + U) ranged from 16% to 90% (with an average of 58%). These average values are similar to values observed in other fish [25,39,40]. Again, precursors showing similarity with the mature sequence cse-PC-5722-5p stand out because of their low A + U percentage (0.16).
The minimum folding free energy value is an important parameter for evaluating the potential secondary structure of RNAs. miRNAs have lower values than some RNAs, but not others. Therefore, additional criteria are required, such as the minimum folding free energy index. This parameter ranged from 0.70 to 3.70 (with an average of 1.09) in our results. Studies in different fish have shown similar or lower values, such as Huang et al.'s study in eleven fish species (range: 0.58 to 0.91, average: 0.71) [39]; C. semilaevis: (range: 0.51 to 1.31, average: 0.90) [41]; and in Boleophthalmus pectinirostris (range: 0.51 to 1.25, average: 0.90) [40]. According to the results obtained by Zhang et al. 's [42], a majority of miRNA precursors have values over 0.85, allowing miRNAs to be discriminated from other coding and non-coding RNA. In our case, 80% of the results had values higher than 0.85, indicating that the predicted miRNAs are true positives and not other types of RNA.
3.2. Phylogenetic Analysis of miRNAs
The precursors of several miRNAs (let-7, miR-130, miR-153, miR-205, miR-301, miR-454, and miR-460) were compared with available stem-loop sequences of the same family from other fish (Supplementary data 2 and Figure 2).
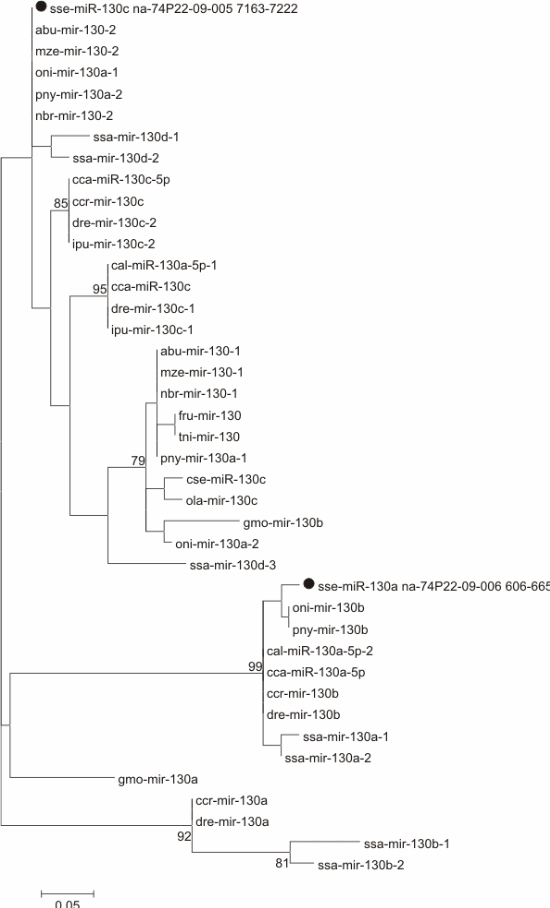
Figure 2 Maximum likelihood phylogenetic tree for precursor sequence miR-130 from Solea senegalensis (marked with a black dot) and other fish. Species abbreviations: Abu: Astatotilapia burtoni, Cca (sequences from [36]) and Ccr: Cyprinus carpio, Dre: Danio rerio, Eel: Electrophorus electricus, Fru: Fugu rubripes, Gmo: Gadus morhua, Hhi: Hippoglossus hippoglossus, Ipu: Ictalurus punctatus, Mze: Metriaclima zebra, Nbr: Neolamprologus brichardi, Oni: Oreochromis niloticus, Ola: Oryzias latipes, Pol: Paralichthys olivaceus, Pny: Pundamilia nyererei, Ssa: Salmo salar, Tni: Tetraodon nigroviridis.
The miRNA family with the largest number of sequences was let-7 with 215; this includes sequences for closely-related mature sequences (from let-7a to let-7j). Although most bootstrap values were low (below 70), sequences usually formed clusters composed of sequences of the only same type (e.g. let-7d, let-7f, let-7g, and let-7i). This points to a high degree of evolutionary conservation. Among the sequences showing similarity to let-7 in S. senegalensis, the sequence sse-let-7a formed a group with let-7a miRNAs, including one Pleuronectiform (C. semilaevis, cse-let-7a-5p). The sequence sse-let-7e was part of a cluster with good bootstrap support (88) containing all let-7e sequences, including cse-let-7e-5p from C. semilaevis, plus two sequences from C. alburnus (cal-let-7-5p-22 and cal-let-7-5p-11).
The tree for mir-130 (Fig. 2) did not show a clear clustering among related sequences (mir-130a, mir-130b, and mir-130c). Sequence sse-miR-130a formed a group displaying good bootstrap support (99) with a combination of mir-130a and mir-130b sequences. On the other hand, sse-miR-130c clustered with low bootstrap support (63) with several Cichliformes and one Cypriniform (mir-130 and mir-130a sequences).
Among the mir-153 sequences available for fish, there were three types of closely-related mature sequences: mir-153a, mir-153b, and mir-153c. Most mir-153a sequences, apart from ssa-mir-153a-1 and ssa-mir-153a-2, formed a cluster (with a bootstrap value of 81). For mir-153b and mir-153c there was not a clear separation, and sequence sse-miR-153b clustered with 153b and 153c miRNAs.
Two main clusters (with a bootstrap value of 70) were observed for mir-301, one containing mostly mir-301a sequences and the other mostly mir-301b and mir-301c sequences. The sequence sse-miR-301b was positioned close to mir-301b sequences, including cse-miR-301b-5p from C. semilaevis.
No clear clustering was observed for sse-miR-460 with miR-460 sequences, despite having a sequence for C. semilaevis, a Pleuronectiformes species. Nevertheless, well-supported clusters were observed for Cichliformes and Cypriniformes species (as well as a Siluriformes species). Two other miRNAs, miR-205 and miR-454, did not show a clear clustering.
In addition, the precursor sequences of two miRNAs, miR-467 and miR-8915, appeared multiple times and were aligned. Three of the miR-467 precursor sequences were identical (sse-miR-467g-1, sse-miR-467g-2, and sse-miR-467g-4), and the others ranged in length from 64 to 105 nt. When the maximum likelihood tree was obtained (data not shown), two groups were observed (with a bootstrap value of 100), one with the identical sequences and the other with the remaining ones. The precursor sequences for sse-miR-8915 were almost identical, with one sequence being 1 nt shorter (sse-miR-8915-1) and the other showing a substitution (sse-miR-8915-4, position 15, with an A instead of a G). This strongly suggests that miR-467 and miR-8915 genes might have suffered gene duplications during evolution, and that there are several loci producing the same mature sequence.
The observation that some miRNAs are considerably conserved among teleosts suggests that the information obtained in S. senegalensis might be transferable to other fish species.
3.3. Target Prediction
Of the 59514 sequences present in the reference transcriptome for S. senegalensis, 12387 were putative targets according to miRanda, with some of them appearing up to seven times. Among the 34 unique sequences with similarity to mature miRNAs that showed potential targets, sse-miR-3584 and sse-miR-4698 produced the most results (with more than twice as many potential targets as the next miRNA), with 3234 and 2748 hits, respectively.
The top ten most frequent gene ontology terms (out of a total of 5857) are presented in Table 2. The predicted targets are implicated in a great variety of processes such as ion binding and nucleic acid binding, or are membrane-related. The sequences showing the highest score for each of the 34 mature sequences are shown in table S2. For example, sse-PC-15046 (solea_v4.1_unigene226900) targets a gene in D. rerio [43] that encodes an E3 ubiquitin ligase that targets a receptor in the Notch signalling pathway, which is involved in a wide range of developmental processes. The sse-miR-153b mature sequence targets a gene of the odorant receptor gene family; these genes exhibit several transmembrane domains and show great diversity in teleost fish [44]. The number of predicted target genes observed implies that miRNAs in this species modulate a wide range of processes. This information is essential for understanding development and other physiological processes, such as larval and sex development as well as disease resistance, and it will contribute to improving the aquaculture process in this and other species. Nevertheless, these results need to be taken with caution. Several factors might have a considerable impact on the rate of false positive results: (a) the lack of information on gene structure without properly defined 3' UTR regions, as is the case for most S. senegalensis genes, and (b) the difficulty of predicting and investigating the precise way that a miRNA and its mRNA target pair interact [35]. Experimental validation is required to confirm the predictions obtained here.
Table 2 Ten most frequent gene ontology terms found for the targets of 34 putative miRNAs in Solea senegalensis.
| Number
|
GO term
|
Name
|
| 1053
|
GO:0008270
|
zinc ion binding
|
| 932
|
GO:0005524
|
ATP binding
|
| 898
|
GO:0016021
|
integral component of membrane
|
| 831
|
GO:0005634
|
nucleus
|
| 691
|
GO:0005622
|
intracellular
|
| 573
|
GO:0003676
|
nucleic acid binding
|
| 432
|
GO:0003700
|
DNA-binding transcription factor activity
|
| 416
|
GO:0000166
|
nucleotide binding
|
| 396
|
GO:0005509
|
calcium ion binding
|
| 380
|
GO:0003677
|
DNA binding
|
4. Conclusions
In this work, an in silico approach was used to identify miRNAs in S. senegalensis from sequences of a BAC library. Using the mature sequences available at miRBase and other sources, 45 groups of sequences showed similarity to mature miRNAs. Of these, 19 were assigned to Senegalese Sole chromosomes using BAC-FISH data. Phylogenetic trees showed different levels of support for clustering among types of miRNAs, and S. senegalensis sequences usually were close to other Pleuronectiformes. Thirty-four unique mature sequences targeted 12387 sequences from an S. senegalensis reference transcriptome. This suggests that miRNAs might modulate a wide range of processes in this species. These results might be helpful for understanding processes such as sexual differentiation, larval development, and disease susceptibility, which hamper the commercial utilization of this species in aquaculture. The phylogenetic conservation of some miRNA implies that the information obtained in the Senegalese sole might be transferable to other aquaculture fish species. Nevertheless, experimental validation by RT-PCR, northern blotting, or microarrays is required.
Additional Materials
The following additional materials are uploaded at the page of this paper.
1. Table S1. Potential miRNAs identified in Solea senegalensis BAC sequences. Group numbers (col. 1) with two integers separated by a dot indicate multiple mature miRNAs matching to the same or almost the same position. When a miRNA name (col. 3) corresponds to a group name (col. 2) this means that this sequence (col. 4) is the 5’ or 3’ arm of the other mature sequence. Sequence names (col. 4) contain the following four items of information separated by dashes: chromosome number (99: hybridization to multiple chromosomes, na: no hybridization); BAC name (plate number: first two characters, and plate position: last three characters); sequencing batch; and contig number.
2. Table S2. Sequences from Solea senegalensis reference transcriptome (v4.1) showing the highest score as targets of 34 putative miRNAs.
3. Supplementary data 1. Secondary structures for the 124 miRNA precursors found for Solea senegalensis. For miRNA group identification numbers see table S1.
4. Supplementary data 2. Maximum likelihood phylogenetic trees in Newick format for precursor sequences let-7, miR-130, miR-153, miR-250, miR-301, miR-454 and miR-460, from Solea senegalensis and other fish. Species abbreviations: Abu: Astatotilapia burtoni, Cca (sequences from Huang et al. Gene Reports, 2017, 8: 30-36) and Ccr: Cyprinus carpio, Dre: Danio rerio, Eel: Electrophorus electricus, Fru: Fugu rubripes, Gmo: Gadus morhua, Hhi: Hippoglossus hippoglossus, Ipu: Ictalurus punctatus, Mze: Metriaclima zebra, Nbr: Neolamprologus brichardi, Oni: Oreochromis niloticus, Ola: Oryzias latipes, Pol: Paralichthys olivaceus, Pny: Pundamilia nyererei, Ssa: Salmo salar, Tni: Tetraodon nigroviridis.
5. Supplementary data 3. Target prediction results from Solea senegalensis reference transcriptome (v4.1) and 34 putative miRNAs using miRanda. Unigen_id: unigene identification code, Functional_description: functional description (see Solea-DB for details), GO_terms: gene ontology terms, mature_miRNA: name of the mature miRNA, Tot Score: total score, Tot Energy: total energy, Max Score: maximum score, Max Energy: maximum energy, Len1: mature miRNA sequence length, Len2: length of the reference sequence examined, Positions: potential target sites.
Author Contributions
AAP, DRT and LR conceived and designed the analysis and AAP and DRT developed the pipeline. LR, IC and TL provided guidance and support. MER, SPB, EGS, MAM and AGA contributed to BAC-FISH data collection and analysis. AAP, DRT and LR wrote the manuscript. All the authors have read and contributed to the manuscript.
Funding
This study has been supported by the Spanish Ministerio de Ciencia e Innovación MICINN-FEDER (project AGL2014-51860-C2-1-P awarded to Laureana Rebordinos).
Competing Interests
The authors have declared that no competing interests exist.
References
- Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and function. Thromb. Haemost. 2012; 107: 605-610. [CrossRef] [Google scholar] [PubMed]
- Bizuayehu T, Babiak I. MicroRNA in Teleost Fish. Genome Biol Evol. 2014; 6: 1911-1937. [CrossRef] [Google scholar] [PubMed]
- Fjose A, Zhao X. Exploring microRNA functions in zebrafish. N Biotechnol. 2010; 27: 250-255. [CrossRef] [Google scholar] [PubMed]
- Leucci E, Patella F, Waage J, Holmstrom K, Lindow M, Porse B et al. microRNA-9 targets the long non-coding RNA MALAT1 for degradation in the nucleus. Sci Rep. 2013; 3: 2535. [CrossRef] [Google scholar] [PubMed]
- Bartel D. MicroRNAs: target recognition and regulatory functions. Cell. 2009; 136: 215-233. [CrossRef] [Google scholar] [PubMed]
- Griffiths-Jones S, Saini H, van Dongen S, Enright A. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008; 36: D154-D158. [CrossRef] [Google scholar] [PubMed]
- Lee R, Feinbaum R, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993; 75: 843-854. [CrossRef] [Google scholar] [PubMed]
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern-formation in C. elegans. Cell. 1993; 75: 855-862. [CrossRef] [Google scholar] [PubMed]
- Wang J, Yang L, Zhang J, Gong J, Wang Y, Zhang C et al. Effects of microRNAs on skeletal muscle development. Gene. 2018; 668: 107-113. [CrossRef] [Google scholar] [PubMed]
- Deb B, Uddin A, Chakraborty S. miRNAs and ovarian cancer: An overview. J. Cell. Physiol. 2018; 233: 3846-3854. [CrossRef] [Google scholar] [PubMed]
- Megha S, Basu U, Kav N. Regulation of low temperature stress in plants by microRNAs. Plant Cell Environ. 2018; 41: 1-15. [CrossRef] [Google scholar] [PubMed]
- Rasal K, Nandanpawar P, Swain P, Badhe M, Sundaray J, Jayasankar P. MicroRNA in aquaculture fishes: a way forward with high-throughput sequencing and a computational approach. Rev Fish Biol Fish. 2016; 26: 199-212. [CrossRef] [Google scholar]
- Herkenhoff M, Oliveira A, Nachtigall P, Costa J, Campos V, Hilsdorf A et al. Fishing into the microRNA transcriptome. Front Genet. 2018; 9: 88. [CrossRef] [Google scholar] [PubMed]
- Díaz-Ferguson E, Cross I, Barrios M, Rebordinos L. Genetic relationships among populations of the Senegalese Sole Solea senegalensis in the Southwestern Iberian Peninsula detected by mitochondrial DNA–restriction fragment length polymorphisms. T Am Fish Soc. 2007; 136: 484-491. [CrossRef] [Google scholar]
- Imsland A, Foss A, Conceicao L, Dinis M, Delbare D, Schram E et al. A review of the culture potential of Solea solea and S. senegalensis. Rev Fish Biol Fish. 2003; 13: 379-407. [CrossRef] [Google scholar]
- Morais S, Aragao C, Cabrita E, Conceicao L, Constenla M, Costas B et al. New developments and biological insights into the farming of Solea senegalensis reinforcing its aquaculture potential. Rev Aquacul. 2016; 8: 227-263. [CrossRef] [Google scholar]
- Benzekri H, Armesto P, Cousin X, Rovira M, Crespo D, Alejandro Merlo M et al. De novo assembly, characterization and functional annotation of Senegalese sole (Solea senegalensis) and common sole (Solea solea) transcriptomes: integration in a database and design of a microarray. BMC Genomics. 2014; 15: 952. [CrossRef] [Google scholar] [PubMed]
- Portela-Bens S, Merlo M, Rodríguez M, Cross I, Manchado M, Kosyakova N et al. Integrated gene mapping and synteny studies give insights into the evolution of a sex proto-chromosome in Solea senegalensis. Chromosoma. 2017; 126: 261-277. [CrossRef] [Google scholar] [PubMed]
- Taboada X, Rey M, Bouza C, Viñas A. Cytogenomic analysis of several repetitive DNA elements in turbot (Scophthalmus maximus). Gene. 2018; 644: 4-12. [CrossRef] [Google scholar] [PubMed]
- Benitez-Dorta V, Caballero M, Betancor M, Manchado M, Tort L, Torrecillas S et al. Effects of thermal stress on the expression of glucocorticoid receptor complex linked genes in Senegalese sole Solea senegalensis: Acute and adaptive stress responses. Gen Comp Endocrinol. 2017; 252: 173-185. [CrossRef] [Google scholar] [PubMed]
- Campos C, Sundaram A, Valente L, Conceição L, Engrola S, Fernandes J. Thermal plasticity of the miRNA transcriptome during Senegalese sole development. BMC Genomics. 2014; 15: 525. [CrossRef] [Google scholar] [PubMed]
- Li Y, Liu S, Qin Z, Waldbieser G, Wang R, Sun L et al. Construction of a high-density, high-resolution genetic map and its integration with BAC-based physical map in channel catfish. DNA Res. 2015; 22: 39-52. [CrossRef] [Google scholar] [PubMed]
- García-Cegarra A, Merlo M, Ponce M, Portela-Bens S, Cross I, Manchado M et al. A Preliminary Genetic Map in Solea senegalensis (Pleuronectiformes, Soleidae) Using BAC-FISH and Next-Generation Sequencing. Cytogenet Genome Res. 2013; 141: 227-240. [CrossRef] [Google scholar] [PubMed]
- Huang Y, Gong W, Ren H, Xiong J, Gao X, Sun X. Identification of the conserved and novel microRNAs by deep sequencing and prediction of their targets in Topmouth culter. Gene. 2017; 626: 298-304. [CrossRef] [Google scholar] [PubMed]
- Sha Z, Gong G, Wang S, Lu Y, Wang L, Wang Q et al. Identification and characterization of Cynoglossus semilaevis microRNA response to Vibrio anguillarum infection through high-throughput sequencing. Dev. Comp. Immunol. 2014; 44: 59-69. [CrossRef] [Google scholar] [PubMed]
- Huang Y, Yang Y, Gao X, Ren H, Xiong J, Sun X. Genome-wide identification and characterization of microRNAs and target prediction by computational approaches in common carp. Gene Reports. 2017; 8: 30-36. [CrossRef] [Google scholar]
- Bizuayehu T, Lanes C, Furmanek T, Karlsen B, Fernandes J, Johansen S et al. Differential expression patterns of conserved miRNAs and isomiRs during Atlantic halibut development. BMC Genomics. 2012; 13: 11. [CrossRef] [Google scholar] [PubMed]
- Fu Y, Shi Z, Wu M, Zhang J, Jia L, Chen X. Identification and differential expression of microRNAs during metamorphosis of the Japanese Flounder (Paralichthys olivaceus). Plos One. 2011; 6: e22957. [CrossRef] [Google scholar] [PubMed]
- Zhang B, Zhang J, Sun L. In-depth profiling and analysis of host and viral microRNAs in Japanese flounder (Paralichthys olivaceus) infected with megalocytivirus reveal involvement of microRNAs in host-virus interaction in teleost fish. BMC Genomics. 2014; 15: 878. [CrossRef] [Google scholar] [PubMed]
- Robledo D, Paula Martin A, Antonio Alvarez-Dios J, Bouza C, Gomez Pardo B, Martinez P. First characterization and validation of turbot microRNAs. Aquaculture. 2017; 472: 76-83. [CrossRef] [Google scholar]
- Merlo M, Iziga R, Portela-Bens S, Cross I, Kosyakova N, Liehr T et al. Analysis of the histone cluster in Senegalese sole (Solea senegalensis): evidence for a divergent evolution of two canonical histone clusters. Genome. 2016; 60: 441-453. [CrossRef] [Google scholar] [PubMed]
- Mathelier A, Carbone A. MIReNA: finding microRNAs with high accuracy and no learning at genome scale and from deep sequencing data. Bioinformatics. 2010; 26: 2226-2234. [CrossRef] [Google scholar] [PubMed]
- Hofacker I, Fontana W, Stadler P, Bonhoeffer L, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh Chem. 1994; 125: 167-188. [CrossRef] [Google scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 2018; 35: 1547-1549. [CrossRef] [Google scholar] [PubMed]
- Enright A, John B, Gaul U, Tuschl T, Sander C, Marks D. MicroRNA targets in Drosophila. Genome Biol. 2004; 5: R1. [CrossRef] [Google scholar] [PubMed]
- Xiong P, Hulsey C, Meyer A, Franchini P. Evolutionary divergence of 3’ UTRs in cichlid fishes. BMC Genomics. 2018; 19: 433. [CrossRef] [Google scholar] [PubMed]
- Thatcher E, Bond J, Paydar I, Patton J. Genomic organization of zebrafish microRNAs. BMC Genomics. 2008; 9: 253. [CrossRef] [Google scholar] [PubMed]
- Guo L, Lu Z. The fate of miRNA* strand through evolutionary analysis: implication for degradation as merely carrier strand or potential regulatory molecule? Plos One. 2010; 5: 1-9. [CrossRef] [Google scholar] [PubMed]
- Huang Y, Zou Q, Ren H, Sun X. Prediction and characterization of microRNAs from eleven fish species by computational methods. Saudi J Biol Sci. 2015; 22: 374-381. [CrossRef] [Google scholar] [PubMed]
- Gong W, Huang Y, Xie J, Wang G, Yu D, Sun X. Genome-wide identification of novel microRNAs from genome sequences using computational approach in the mudskipper (Boleophthalmus pectinirostris). Russ J Bioorganic Chem. 2017; 43: 397-408. [CrossRef] [Google scholar]
- Huang Y, Yang Y, Sun X. Genome-wide identification of microRNAs and their target genes in Cynoglossus semilaevis using computational approach. Gene Reports. 2016; 4: 235-243. [CrossRef] [Google scholar]
- Zhang B, Pan X, Cox S, Cobb G, Anderson T. Evidence that miRNAs are different from other RNAs. Cell Mol Life Sci. 2006; 63: 246-254. [CrossRef] [Google scholar] [PubMed]
- Zhang C, Li Q, Jiang Y. Zebrafish mib and mib2 are mutual e3 ubiquitin ligases with common and specific delta substrates. J Mol Biol. 2007; 366: 1115-1128. [CrossRef] [Google scholar] [PubMed]
- Alioto T, Ngai J. The odorant receptor repertoire of teleost fish. BMC Genomics. 2005; 6: 173. [CrossRef] [Google scholar] [PubMed]

