

Innate Immune Determinants of Graft-Versus-Host Disease and Bidirectional Immune Tolerance in Allogeneic Transplantation
Anouk A. J. Hamers 1, 2, 3, 4 ![]() , Sunil K. Joshi 1, 2, 3, 4
, Sunil K. Joshi 1, 2, 3, 4 ![]() , Asha B. Pillai 1, 2, 3, 4, 5 *
, Asha B. Pillai 1, 2, 3, 4, 5 * ![]()
- Department of Pediatrics, Division of Hematology / Oncology and Bone Marrow Transplantation, University of Miami Miller School of Medicine, Miami, FL, USA
- Batchelor Children's Research Institute, University of Miami Miller School of Medicine, Miami, FL, USA
- Department of Microbiology & Immunology, University of Miami Miller School of Medicine, Miami, FL, USA
- Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL, USA
- Holtz Children’s Hospital, University of Miami Miller School of Medicine, Miami, FL, USA.
* Correspondence: Asha B. Pillai ![]()
Academic Editor: Jean Kwun
Special Issue: Multiple Aspects of Transplant Tolerance – Mechanisms, Strategies, and Barriers
Received: September 29, 2018 | Accepted: January 28, 2019 | Published: January 31, 2019
OBM Transplantation 2019, Volume 3, Issue 1 doi: 10.21926/obm.transplant.1901044
Recommended citation: Hamers AAJ, Joshi SK, Pillai AB. Innate Immune Determinants of Graft-Versus-Host Disease and Bidirectional Immune Tolerance in Allogeneic Transplantation. OBM Transplantation 2019; 3(1): 044; doi:10.21926/obm.transplant.1901044.
© 2019 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
The success of tissue transplantation from a healthy donor to a diseased individual (allo-transplantation) is regulated by the immune systems of both donor and recipient. Developing a state of specific non‐reactivity between donor and recipient, while maintaining the salutary effects of immune function in the recipient, is called “immune (transplantation) tolerance”. In the classic early post‐transplant period, minimizing bidirectional donor ←→ recipient reactivity requires the administration of immunosuppressive drugs, which have deleterious side effects (severe immunodeficiency, opportunistic infections, and neoplasia, in addition to drug-specific reactions and organ toxicities). Inducing immune tolerance directly through donor and recipient immune cells, particularly via subsets of immune regulatory cells, has helped to significantly reduce side effects associated with multiple immunosuppressive drugs after allo‐transplantation. The innate and adaptive arms of the immune system are both implicated in inducing immune tolerance. In the present article, we will review innate immune subset manipulations and their potential applications in hematopoietic stem cell transplantation (HSCT) to cure malignant and non-malignant hematological disorders by inducing long-lasting donor ←→ recipient (bidirectional) immune tolerance and reduced graft-versus-host disease (GVHD). These innate immunotherapeutic strategies to promote long‐term immune allo-transplant tolerance include myeloidderived suppressor cells (MDSCs), regulatory macrophages, tolerogenic dendritic cells (tDCs), Natural Killer (NK) cells, invariant Natural Killer T (iNKT) cells, gamma delta T (γδ‐T) cells and mesenchymal stromal cells (MSCs).
Keywords
Innate immune cells; central tolerance; peripheral tolerance; allo-transplantation; Graft-versus-Host Disease; rejection; thymic selection; regulatory T Cells; immunosuppression; mixed chimerism
1. Introduction
Clinical allograft rejection was first reported by Earl C. Padgett in 1932, when he applied unrelated skin grafts to treat burn patients. Padgett noted that, whereas skin grafts derived from donors unrelated to the recipients were rejected within a short time, grafts deriving from close relatives were not immediately rejected by the transplant recipient. This was the first observation of an effect of genetic compatibility between donor and recipient on graft survival, a phenomenon mechanistically explained when the immunological basis of allo-rejection was later elucidated by Gibson and Medawar in 1943 [1,2,3]. In 1948, Medawar and colleagues made the key observation that cellular components of the recipient immune system are responsible for the rejection of the graft and that this phenomenon was subject to some form of immunologic “memory” [4,5,6,7]. Medawar’s observation was complemented in 1949 by Burnet’s theory that the immune system can discriminate between foreign antigens (i.e. "non-self” antigens) and antigens derived from one’s own body (i.e. "self” antigens) [1,3]. In 1957, E. Donnall Thomas documented the first human allogeneic hematopoietic stem cell transplantation (allo-HSCT) procedure followed by much of his developmental work on bone marrow and blood-derived HSCT [8,9,10].
The advent of immunosuppressive drugs led to markedly improved success in preventing graft rejection. In 1972, Cyclosporine A (CsA) was found to be lymphocyte immunosuppressive and was approved in 1980 for use in allo-transplantation [11,12]. However, CsA and other existing immunosuppressive drugs have deleterious side effects including renal, hepatic, cardiovascular and metabolic dysfunction, opportunistic infections, and secondary malignancies. To improve the quality of life for patients undergoing allo-HSCT, new strategies are evolving to reduce or eliminate the use of immunosuppressive drugs post-HSCT (reviewed in [13]).
Non-reactivity between donor and recipient immune systems is defined in specific ways, each with specific clinical correlates (Figure 1A). Lack of responsiveness between donor and recipient without ability to respond to third party antigens (non-specific non-responsiveness) is termed ‘anergy’. Anergy is a form of generalized immune suppression and can clinically associate with lack of robust responses to exogenous or endogenous pathogens. Transplant tolerance is distinct from anergy in that it is donor ←→ recipient alloantigen-specific, with maintenance of reactivity to third party antigens and, importantly, pathogens (reviewed in [7,14,15,16]).
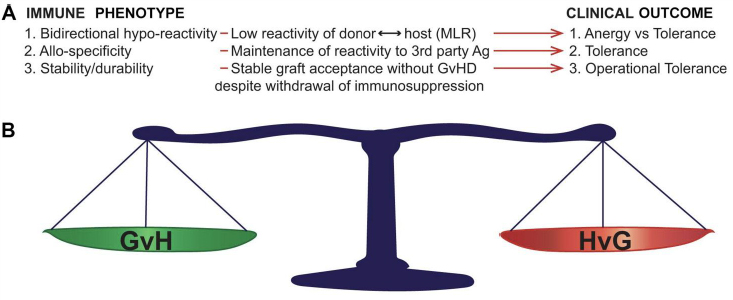
Figure 1 Bidirectional immune features required for operational tolerance (OT) in allo-transplantation. A: Immune correlates and associated clinical outcomes. After OT induction in either HSCT or solid organ transplantation, specific immune phenomena (Immune Phenotype) correlate with specific measurable correlates. The specific clinical outcome associated with each phenotype is defined on the right of the figure (Clinical Outcome). B: OT requires bidirectionality donor recipient immune tolerance. Not only must OT fulfil specific immune criteria in the absence of immune suppression, but this stable long-range balance must be maintained in both the GvH and HvG directions in order to achieve durable graft acceptance without GVHD. MLR, mixed leukocyte reaction; GvH, graft-versus-host reactivity (associated with graft-versus-host disease); HvG, host-versus-graft reactivity (associated with graft rejection).
2. Strategies to Induce Long-Term Immune Tolerance after Allo-transplantation
The distinction between anergy and tolerance (Figure 1) is not merely academic, as multiple translational studies have shown the relevance of antigen-specific hyporeactivity between donor and recipient to the maintenance of a healthy graft without graft-versus-host disease (GVHD), while maintaining host resistance to exogenous pathogens [14,17,18,19]. Once antigen-specific tolerance is achieved, the next step is insurance of a stability of this tolerance. This is because maintaining a transplant recipient on lifelong immunosuppression is known to greatly compromise quality of life and increase the risk of transplant-related death from serious infections and drug-associated acute and chronic complications. When (alloantigen-specific) transplant tolerance can be maintained in the absence of drug-induced immunosuppression, this is known as ‘operational tolerance’. To achieve operational tolerance (OT) is the “holy grail” in allo-transplantation and is thus a major focus of preclinical and translational research. Importantly, OT requires bidirectionality (stable maintenance of host-versus-graft and graft-versus-host antigen-specific hyporeactivity) (Figure 1A) in order to be clinically meaningful, as imbalance of either results in graft rejection or GVHD.
3. Mixed Chimerism as a Means of Operational Tolerance Induction
One well-studied and translated mechanism for OT induction which does not relate solely on individual immune subsets is the induction of mixed chimerism. This involves rebooting the immune system of the transplant recipient by inducing tolerance to the hematopoietic stem cell (HSC)-derived immune components of the graft donor, and vice versa. After successful OT induction in allo-HSCT, HSCs from the donor and recipient can stably coexist in a patient via induction of host-versus-graft (HvG) and graft-versus-host (GvH) tolerance, also termed “bidirectional immune tolerance” (Figure 1B). This state is characterized by stable co-existence of multilineage hematopoiesis of both recipient and donor, termed ‘stable mixed chimerism’ [20,21,22]. Additionally, the introduction of an immune system derived from the transplanted HSCs that matches the transplanted organ can result in ongoing thymic deletion of recipient T cell clones capable of rejecting the graft, as well as abrogation of donor T cell clones capable of inducing GVHD. This has successfully achieved solid organ tolerance in human clinical trials. However, such “mixed chimerism tolerogenic protocols” include all of the risks and toxicities associated with HSCT, including short- and long-term toxicity of HSCT conditioning regimens ,[21,23,24]. In this review, we will focus on the roles of innate immune cell subsets of both the donor and recipient immune system in GVHD and donor ←→ recipient transplantation tolerance after HSCT. We will also discuss the potential therapeutic applications of these cells to cure malignant and non-malignant hematological disorders by inducing long-lasting immune tolerance and reducing GVHD.
4. Innate Immune Transplant Tolerance
The innate immune system forms the first line of defense against foreign pathogens. Innate immune mechanisms are critical to consider in effecting and maintaining transplantation tolerance across histocompatibility barriers. By definition, these mechanisms are not restricted by major histocompatibility complex (MHC) antigens or, in humans, Human Leukocyte Antigens (HLA). Thus, these components can be harnessed to maintain immune tolerance following HLA-incompatible transplants, as we have shown in murine models of MHC-mismatched HSCT [25,26,27,28].
4.1 Innate Myeloid Populations
MDSCs, regulatory macrophages, and tolerogenic DCs have all been implicated in allo-HSCT transplant tolerance.
4.1.1 Myeloid-Derived Suppressor Cells (MDSCs)
Nomenclature and Phenotype. MDSCs, expand and recover rapidly after allo-HSCT [29,30] and can contribute directly to the immunosuppressive tumor microenvironment by dampening T effector cell responses (reviewed in [31,32]). While this immunosuppressive function may be detrimental in the immune response to cancer, it can be instrumental in achieving transplant tolerance after allo-HSCT [27,28,29,30], particularly for non-malignant disorders in which anti-tumor activity is not a consideration. MDSCs constitute a heterogeneous population which can be classified into two broad sub-categories: granulocytic/neutrophilic MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs) [33,34,35]. Murine PMN-MDSCs are defined as CD11b+Ly6G+Ly6Clow and M-MDSCs as CD11b+Ly6GnegLy6Chigh. In humans, PMN-MDSCs are defined as CD11b+CD14negCD15+CD66b+LOX-1+ cells and M-MDSCs are defined as CD11b+CD33+CD14+CD15negCD66bnegHLA−DRlow cells [35,36]. Proof of the capacity for T effector cell suppression is obligatory to identify cells as MDSCs. This is because (classical, non-allosuppressive) mature monocytes share many other phenotypic and morphologic features with M-MDSC. The same is true in differentiating neutrophils from PMN-MDSCs; namely, only PMN-MDSCs are known to be allosuppressive [33,34,35,37,38]. In this context, there is some ongoing debate in the field whether MDSCs should be regarded as distinct entities or as monocytes/neutrophils that have been reprogrammed to have immunosuppressive capacity [37,38,39,40,41,42]. Therefore, better characterization of these cells is needed in order to understand how to phenotypically separate them from other mature myeloid cells.
Preclinical Data. In MHC-matched mouse bone marrow transplantation (BMT) studies, donor MDSCs can expand in response to irradiation [43]. Moreover, total MDSC numbers have been correlated to acute GVHD severity in an MHC-mismatched mouse allo-transplant model, and removal of MDSCs aggravates GVHD [44]. In other studies with similar models, donor MDSCs induce allo-HSCT transplantation tolerance by suppressing alloreactive T cells via the enhancement of either arginase-1 or iNOS expression, causing local depletion of L-arginine required for optimal T cell proliferation and survival [45,46]. Interestingly, Wang et al. [44] showed that donor-derived MDSCs from recipients with GVHD were more potent in their inhibition of alloreactive T cell proliferation than donor MDSCs from non-GVHD recipients, suggesting the requirement for an inflammatory immune milieu to optimize MDSC allo-suppressor capacity against GVHD-inducing effector T cells. MacDonald et al. [47] demonstrated that donor MDSCs can suppress GVHD by promoting class-II dependent, interleukin (IL)-10 producing T cells. Indeed, infusion of ex vivo generated human cord blood MDSCs into a murine allo-HSCT model of chronic GVHD results in regulatory T cell (Treg) expansion thereby attenuating GVHD severity [48]. Tregs are important for controlling alloreactive T cell responses to “self” antigens in the non-transplant setting and for maintaining allograft tolerance in both organ transplant and allo-HSCT [49,50]. Although detailed mechanisms for MDSC-dependent Treg expansion in allo-HSCT remain undefined, it may be either contact-dependent via programmed death-ligand 1 (PD-L1) as we have shown [27] or mediated via the paracrine production of regulatory cytokines such as transforming growth factor-beta (TGF-β) and IL-10 [45,51,52,53]. While the above mentioned work has mainly focused on donor MDSCs in MHC-mismatched allo-HSCT after total body irradiation (TBI) in mice, we have shown that recipient MDSCs and Th2 cytokine signaling are both necessary and required to potently suppress GVHD after nonmyeloablative MHC-mismatched HSCT [27] (Figure 2). We demonstrated in a murine GVHD model system that recipient MDSCs have the ability to convert to regulatory myeloid DCs, which in turn augment donor Treg recovery via a PD-L1 dependent mechanism to induce graft-versus-host immune tolerance [27]. Cumulatively, these data point to MDSC as a potential candidate for cellular immunotherapy, both to prevent and to treat GVHD [44,45,46,54]. Toward that end, MDSCs have been expanded in vivo with granulocyte-colony stimulating factor (G-CSF) [44], a synthetic fusion of G-CSF and Flt-3 [47], or IL-33 [55]. In vitro generation of MDSCs can be achieved by culturing BM cells with granulocyte-macrophage colony stimulating factor (GM-CSF) and G-CSF either with or without addition of IL-13 [45,46], or a combination of GM-CSF with IL-6 [56]. Zhou et al. [54] reported a method to use embryonic stem cells for in vitro MDSC differentiation by stimulating these cells with a cocktail of c-kit ligand, vascular endothelial growth factor (VEGF), Flt3 ligand (Flt3L), and thrombopoietin. Notably, relatively high ratios of MDSCs to donor T cells (4:1) appear required for GVHD suppression [45,54]. Human cord blood MDSC infusion into a xenogenic mouse model has shown a reduction in chronic GVHD of skin, lung, and liver [48].

Figure 2 Innate immune networks are a platform for OT induction across histocompatibility barriers. Operational tolerance (OT) is governed through a complex interaction between the innate and adaptive immune systems. The interface between innate (violet) and adaptive (green) is indicated in the figure and represents the bridging of OT across MHC barriers. iNKT, invariant Natural Killer T cell; MØreg, regulatory macrophage; DC, dendritic cell; MDSC, myeloid- derived suppressor cell; MSC, mesenchymal stromal cell; NK, Natural Killer cell; γδ, gamma-delta T cell; Treg, regulatory T cell; Breg, regulatory B cell; Tfr, regulatory follicular helper T cell; Teff, T effector cell.
Clinical Data. Paralleling the data in mice, MDSC numbers have been shown to increase with donor-induced acute GVHD in leukemia patients receiving allo-HSCT [29,30,57]. GM-CSF, G-CSF, and IL-6, factors known to promote MDSC expansion, are highly present post-HSCT [57,58]. CD14+HLA-DRneg/lowIDO+ M-MDSCs, as reported by Mougiakakos et al., are not only significantly increased in acute GVHD patients, but can also suppress T cell proliferation in vitro [57]. Moreover, inhibition of indoleamine 2,3-dioxygenase (IDO) leads to an almost 3-fold increase in T effector cell proliferation and enhanced interferon-gamma (IFN-γ) release [57]. Aside from T cell suppression and in support of our murine findings [27,28], human MDSCs have been shown to enhance recovery of both CD4+ and CD8+ Tregs post allo-HSCT [29,59]. To date, there are no published trials of human MDSC infusions into patients with active GVHD. MDSCs are a rare population in healthy individuals and therefore difficult to isolate. In order to generate enough M-MDSC numbers for infusion, ex vivo expansion procedures are being attempted. These mainly utilize GM-CSF with IL-6 [59], GM-CSF with both IL-6 and VEGF [60], or co-culture with human solid tumor cell lines [60]. Although MDSCs seem promising cellular immunotherapy candidates for inducing and maintaining transplant tolerance, caution is warranted because undifferentiated myeloid subsets are highly adaptable and could theoretically rapidly switch from anti- to pro-inflammatory phenotypes in inflammatory milieus. For example, murine MDSCs are susceptible to inflammasome induction when introduced into a severe inflammatory environment such as that of acute GVHD [61]. Another important therapeutic consideration is that timing of MDSC infusion may be critical to tolerogenesis or to impeding critical anti-viral immunosurveillance [62].
4.1.2 Monocytes and Macrophages
Nomenclature and Phenotype. Macrophages and their (monocyte) precursors rapidly migrate into inflamed tissues in response to injury. Macrophages are the major cell type infiltrating chronically rejected allo-grafts [63] and constitute a phenotypically heterogeneous and remarkably plastic population which can differentiate into a wide spectrum of functional subsets depending upon the tissue microenvironment [63,64]. In response to different stimuli, infiltrating monocytes preferentially differentiate into “classically activated” or “alternatively activated” macrophages with divergent functions. Classically activated macrophages (M1 macrophages) develop in response to IFN-γ and lipopolysaccharide (LPS), secrete high levels of pro-inflammatory cytokines, and typically promote a Th1 response [65]. These pro-inflammatory M1 macrophages express high levels of arginase-2, inducible nitric oxide synthase (iNOS), tumor necrosis factor-alpha (TNF-α), IL-1, IL-6, and CD86 (reviewed in [66]). In contrast, exposure to Th2-polarizing cytokines leads to the development of alternatively activated M2 macrophages. M2 macrophages are typically regulatory and can be subdivided into M2a, M2b, and M2c subsets, each identifiable by their surface markers and cytokine release profile in response to inflammatory stimuli (reviewed in [66,67]). All three M2 subsets share the capacity for potent IL-10 secretion. M2a macrophages typically express arginase-1, CD206, CD209, FcεR, and the scavenger receptor CD163 and suppress tissue inflammation [68,69]. The M2b subset, induced by co-ligation of macrophage FcRs by IgG complexes coupled with toll-like receptor (TLR), CD40 or IL-1R engagement, is immunoregulatory and produces high levels of IL-10, IL-1, IL-6 and TNF-α [66]. M2c tissue repair macrophages are induced with IL-10, express cell-surface SLAM, CD206, and FIZZ1, and produce high amounts of IL-10 and TGF-β [66].
Preclinical Data. Allograft-infiltrating macrophages can cause allograft injury/rejection, tissue remodeling, or graft-site immunoregulation via diverse mechanisms. Thus macrophages can have either protective or detrimental functions in the allo-transplant setting, dependent upon immune phenotypes. For example, M1 macrophages are associated with T cell independent acute allograft rejection. In acute GVHD the tissue repair functions of recipient M2 macrophages contribute to resolving inflammation. However, in chronic GVHD these same M2 mechanisms can contribute to fibrosis and delayed allograft failure. Specific experimental data follow.
Following myeloablative HSCT, the recipient’s bone marrow (BM) microenvironment is severely damaged due to the cellular stress and tissue injury associated with the pre-conditioning regimen, including adenosine triphosphate (ATP) and Th1 cytokine release, apoptosis of the supporting cells, and microbial antigen leakage from the gut. The latter in turn activates M1 macrophages (reviewed in [65]). M1 macrophages are associated with T cell-independent acute allograft rejection in a murine model of MHC-mismatched allo-HSCT [70,71,72]. Recipient M1 macrophages can exhibit potent cytotoxicity against the donor BM via the TLR4/toll-interleukin-1 receptor domain-containing adapter inducing interferon-β (TRIF) [71]. TLR4 activation has been identified as a barrier to transplant tolerance (reviewed in [65]). TLR4 recognizes LPS, which can leak from the gut in response to radiation-associated conditioning regimens (reviewed in [73]).
In acute GVHD the tissue repair functions of recipient M2 macrophages contribute to resolving inflammation. In contrast to M1 macrophage TLR4 induction of HSCT allograft rejection [71], Imado et al. [74] showed a protective role for host macrophages with regards to acute GVHD. In agreement with Imado et al. [74], the Merad group has shown that reduction of recipient macrophage numbers results in increased donor T cell expansion, thereby exacerbating acute GVHD after allo-HSCT [75]. Conversely, when recipients were given CSF-1 therapy pre-HSCT (a treatment which expands M2 macrophages), donor T cell expansion was reduced and GVHD morbidity and mortality improved [75]. Recipient macrophages persisting post allo-HSCT are capable of engulfing donor allogeneic T cells directly [75], thus regulating or preventing GVHD. However, in chronic GVHD these same M2 macrophage subsets can be detrimental; in mouse models of chronic GVHD, recipient M2 macrophages aggravate the disease by attracting myofibroblasts and increasing TGF-β, inducing massive fibrosis [76,77]. M2 macrophage polarization can be induced by ER stress, and macrophages subjected to ER stress have been linked to chronic GVHD [78,79].
Clinical Data. In congruence with the murine preclinical data, an increased ratio of macrophages to total nucleated cells at day 14 post allo-HSCT was associated with delayed engraftment or subsequent graft failure in 32 patients [80]. A genome wide association study (GWAS) in 492 HLA-matched sibling HSCT donor-recipient pairs from Finland and Spain showed that certain single nucleotide polymorphisms (SNPs) in key macrophage functional gene loci such as IL-1R, TNF-α, IL-10, and NFKBIA are associated with acute GVHD [81]. In this same study, IL-1B, IL-23R, TLR9, and NOD2 associated with chronic GVHD. Interestingly, all these factors are known to play a role in the antimicrobial response of macrophages [82], which follows intestinal damage from the conditioning regimen. In addition, it is known that patients with pre-existing intestinal damage pre-HSCT are more likely to develop acute GVHD after HSCT [83]. Recipient dermal macrophages can be found in GVHD lesions and may persist up to one year post allo-HSCT [84]. Plasma CD163, a macrophage scavenger receptor, has been correlated with a higher cumulative incidence of chronic GVHD in a cohort of 167 patients [85]. Increased CD163 with exposure to the anti-inflammatory cytokine IL-10 may contribute to M2 macrophage recruitment and chronic GVHD pathogenesis [86]. To date, the only attempts to apply regulatory macrophages as a cellular immunotherapy are in renal transplant patients. Hutchinson et al. [87] successfully infused 2 patients with donor regulatory macrophages pre-transplantation and maintained graft functionality for 3 years. These regulatory macrophages were generated using M-CSF for 7 days with a 24-hour stimulation with IFN-γ at day 6. This cellular therapy has yet to be tested in allo-HSCT trials. There are some safety concerns with regards to macrophage plasticity after infusion, since these cells change their phenotype based on their specific microenvironment. One could envision mitigating this by timing the infusions pre-HSCT.
4.1.3 Dendritic Cells (DCs)
Nomenclature and Phenotype. As professional antigen presenting cells, DCs are responsible for inducing adaptive immunity essential for priming antigen-specific T cell responses to alloantigens. By regulating these functions, DCs may also promote tolerogenic responses (reviewed in [88,89]). DCs are heterogeneous, with multiple subsets and distinct functions based on their origin, maturation state, and localization. Mature DCs from the blood include plasmacytoid DCs (pDCs), which are CD11cloMHC-IIlo, and myeloid DCs (mDCs) characterized as CD11chiMHC-IIhi. In mice, pDCs can be delineated by their B220 and Siglec-H expression; human pDCs are CD303+CD123+CD304+. Mouse mDCs are recognized as CD24+BDCA-1+Btla+; human mDCs are described as CD11c+CD1c+BDCA-1+ (reviewed in [90]). Designated separately, tolerogenic DCs (tDCs) are a subset of typically immature mDCs with lower MHC-II surface levels, lower expression of co-stimulatory molecules, and a reduced capacity to produce pro-inflammatory cytokines as compared to the mature DCs (reviewed in [89]). In addition to the above mentioned subsets, there are many other types of tissue-resident DCs whose phenotypes and functions have been extensively reviewed elsewhere [90,91].
Preclinical Data. DC maturation states partially define whether they facilitate or inhibit alloresponses in transplantation, with mature DCs classically defined as potent inducers of alloreactivity and thus of organ rejection and GVHD [92,93]. Mature DCs present antigen to allogeneic effector T cells, activating them to attack the allograft and inducing graft rejection (reviewed in [94,95,96]). Most of these rejection studies have been performed in solid organ transplant models (reviewed in [97]). In mice transplanted with MHC-mismatched T cell-depleted BM, surviving recipient cutaneous DCs are sufficient [98] but not required [99] triggers for generation of cutaneous GVHD. Acute GVHD is mostly caused by recipient APCs priming donor T cells in the first few days after allo-HSCT. Like recipient macrophages, recipient DCs can respond to microbial peptides, ATP, and pro-inflammatory cytokines released from the irradiation-damaged gut epithelium and leukocytes. Recipient DC Cathepsin E, an aspartate protease that cleaves bacterial peptides for antigen presentation, regulates DC motility; mice deficient for Cathepsin E are protected from GVHD [100]. Similarly, recipient DC microRNA-155 has been reported as a factor in acute GVHD induction by promoting DC migration towards sites of ATP release [101]. ATP attenuates GVHD by activating the purinergic receptor P2X7R. Recipient mice deficient for P2X7R have decreased GVHD and improved survival associated with reduced donor T effector cell expansion and increased CD4+Foxp3+ Tregs, while donor P2X7R deficiency has no effect on GVHD severity or survival [102,103,104]. Acute GVHD can develop into chronic GVHD when donor mDC MHC-II antigen presentation is impaired, associated with a failure of Treg homeostasis [105]. tDCs can regulate central tolerance by migrating to the thymus, where they may augment negative selection of “self”-/alloreactive thymocytes and the generation of Tregs (reviewed in [106,107,108]). Unlike mature DCs, tDCs do not express the co-stimulatory molecules necessary to fully activate an alloreactive T effector response. Likely for this reason, maintaining long-range immaturity of DCs has been closely associated with the development of OT. tDCs can maintain OT through multiple mechanisms, including driving naïve T cells to hyporeactivity or inducing T effector cell differentiation into Tregs [109,110] (Figure 2). Our group defined a critical role for recipient tolerogenic immature myeloid precursors spared by non-myeloablative pre-HSCT conditioning in maintaining MHC-mismatched OT by generating PD-ligand-expressing immature CD8αneg mDCs, which are both Th2 cytokine signaling-dependent and NKT-dependent [28]. These tDCs augment donor-type antigen-specific Treg proliferation through the PD-1/PD-ligand axis [27,28]. We subsequently defined a means to augment these tDCs in vivo by adding alkylators to pre-HSCT conditioning [27].
The role of pDCs in transplant tolerance is less studied. pDC precursor cells (pre-pDCs) have been shown to facilitate allo-HSCT engraftment through Treg induction [111,112,113]. While data suggest that mature recipient pDCs do not prevent GVHD [114], depletion of pre-pDCs from the donor BM does result in aggravated GVHD [115], supporting a potential tolerogenic role for pDCs in the appropriate milieu. In addition, adoptive transfer of donor pre-pDCs into an MHC-mismatched mouse model of allo-HSCT diminished GVHD [115,116]. Clearer elucidation of the molecular mechanisms by which tDCs and specific subsets of mDCs and pDCs exert their tolerogenic effects will support their application in OT induction.
Clinical Data. In patients undergoing myeloablative HSCT, donor mDCs and pDCs are already detected in peripheral blood one day after allo-HSCT; within 2 weeks post-HSCT, the majority of circulating DCs are of donor origin [117,118]. In contrast, human Langerhans cells survive conditioning regimens and persist long after HSCT [119]. After myeloablative pediatric allo-HSCT, patients with acute GVHD had significantly lower numbers of circulating mDCs and pDCs as compared to non-GVHD patients [120,121]. Chan et al. [122] noted that patients with more severe acute GVHD or extensive chronic GVHD had more recipient pDCs than donor pDCs, while patients with low-grade acute GVHD had complete donor pDC reconstitution. Another study found higher peripheral blood donor pDC levels in chronic GVHD patients versus non-GVHD patients, while recipient pDCs were mostly found in control patients [123]. Also, a high pDC content in the graft was associated with a higher risk of relapse and lower overall survival in patients receiving allo-HSCT [124]. The discrepancies between these studies have been attributed to key differences in conditioning regimens, differences in stem cell source, time of sampling, and pDC identification by flow cytometry, but hey have yet to be meaningfully mechanistically explained.
Infusion of tDCs into a small group of healthy volunteers resulted in an antigen-specific inhibition of T effector cell function [125] and CD8+ Treg induction [126]. Though Phase I clinical trials with autologous tDCs in type 1 diabetic [127], rheumatoid arthritis [128], and Crohn’s disease [129] patients show that tDCs are safely tolerated, to date there are no published trials studying tDC immunotherapy after allo-HSCT. In the past 20 years, extensive efforts have been made to generate maturation-resistant tDCs for this purpose, in vitro. tDCs are typically differentiated and expanded from PBMCs in the presence of GM-CSF and IL-4 with the addition of 1 or more tolerogenic factors [130,131] including IL-10, TGF-β, Vitamin D3, dexamethasone, apoptotic cells, corticosteroids, and/or rapamycin [130,131,132,133,134,135,136,137,138,139,140]. Immature tDCs may be a promising alternative to existing regimens for tolerance induction, but require clinical study in the allo-transplant setting.
4.2 Innate Lymphoid Populations
Innate lymphoid cells are important as both effector and as regulatory cells, controlling tissue inflammation and immune homeostasis. Since these cells are not MHC restricted in the classic sense, but interact closely with MHC-restricted effector and regulatory T cells, they can be thought of as bridging the innate and adaptive arms of immunity. These lymphocytes develop from a common lymphoid progenitor, but lack the stochastic antigen receptor rearrangements seen in conventional MHC-restricted effector and regulatory T cells. NK cells, NKT cells, and γδ-T cells are all innate lymphoid cells capable of contributing to graft rejection, GVHD, graft-versus-tumor effect, or establishment of OT after allo-HSCT.
4.2.1 Natural Killer (NK) Cells
NK cells can directly kill those leukemic target cells that down-regulate their class I MHC to escape CD8 T cell recognition and killing; they are therefore of great interest in cellular immunotherapy and allo-HSCT.
Nomenclature and Phenotype. Human NK cells are defined as CD3negCD56+ and their mouse counterpart is CD3negNK1.1+. Human NK cells can be further subdivided into immature lymph node-/tonsil-homed CD56brightCD16neg cells and mature peripheral blood-/spleen-homed CD56dimCD16+. Mature NK cells are predominantly cytotoxic [141]. In mice, 2 mature subsets can be distinguished based on their CD27 expression [142]. NK cell functionality is regulated by several activating and inhibitory receptors which can be either MHC-I specific or non-specific. In mice, MHC-I specific receptors include the Ly49 family and the C-type lectin molecule CD94. Ly49 receptors recognize the MHC-I isoforms H-2D and H-2K. CD94 is covalently associated with NKG2 family members and binds to the MHC class I molecule. In humans, CD94 can bind to non-classic HLA-E molecules on target cells. The human counterparts of the mouse Ly49 family are the killer immunoglobin-like receptors (KIR). This family of receptors can bind to certain groups of the classical class I molecules HLA-C (KIR2DL1-3, KIR2DS1-2, KIR2DS4), HLA-B (KIR3DL2, KIR2DS4), and HLA-A (KIR3DL1) alleles and the non-classical class I HLA-G (KIR2DL4) (reviewed in [143]). Each NK cell expresses a panoply of inhibitory receptors that bind to “self” and/or “non-self” MHC-I molecules. “Licensed” NK cells express inhibitory receptors to self MHC class I, thus allowing them to be inhibited by class I expression on “self” tissue and preventing autoreactivity [144,145,146,147].
Preclinical Data. Murine NK cells are highly radioresistant, proliferate under lymphopenic conditions, and thus recover early and rapidly after allo-HSCT [148,149]. NK cells are potent cytolytic cells which can induce either allograft rejection or OT; the latter by killing allo-responsive activated DCs and T cells (Figure 2). Recipient “licensed” NK cells do not usually recognize donor MHC-I molecules with their KIR and will therefore lyse class I-expressing donor cells, thus inducing graft rejection after allo-HSCT [150].
However, donor NK cell infusions have also been shown to prevent T cell-mediated GVHD, while maintaining or even amplifying the graft-versus-tumor effect [151,152,153,154,155]. GVHD protection was due to the presence of alloreactive “licensed” NK cells capable of killing activated DCs (Figure 2), as supported by the requirement for Ly49 ligand-mismatch from otherwise MHC-matched donors for GVHD control [152,156] and the fact that Ly49C silencing can induce NK cell alloreactivity [157]. Further emphasizing the importance of NK cell licensing for transplant tolerance, Chow et al. [158] showed that the transfer of intact MHC-I antigen from recipient cells to transplanted donor cells confers a “self” identity on these otherwise foreign cells, giving them the ability to evade detection by recipient NK cells. Once complete donor chimerism was established, transplant cells no longer required host MHC-I protein transfer to survive. Alloreactive NK cells can inhibit direct activation of T cells via perforin-mediated killing of allogeneic donor-derived DCs [156,159,160,161,162]. This does not occur with unprimed T cells [163], supporting that the NK cell effect acts through DC ablation by NK cells. Conversely, non-licensed inflammatory NK cells can contribute to GVHD development by pro-inflammatory Th1 cytokine production [164,165,166].
Clinical Data. In patients undergoing allo-HSCT, NK cells are the first donor-derived lymphocytes to reconstitute [167,168]. Donor NK cells show a robust proliferation immediately after HSCT [169] and multiple studies suggest that higher NK cell numbers reduce GVHD incidence [170,171,172,173,174,175,176,177]. The dominant NK phenotype in the first weeks post-HSCT is CD56brightCD16neg, while healthy individuals have only 10% CD56brightCD16neg and 90% CD56dimCD16++ NK cells [178,179,180]. In most patients, the relative NK cell expansion normalizes 12 months post-HSCT [168]. Levels of recipient versus donor immune subset chimerism may play a role in allo-HSCT. Breuer et al. [181] identified 3 risk groups for leukemia relapse in children after reduced intensity or myeloablative HSCT, based on recipient chimerism (RC): 1) a high-risk group with both T cell and NK cell RCs above 90%; 2) an intermediate-risk group with a T cell RC above 50% and NK cell RC below 90%; and 3) a low-risk group with a T cell RC below 50%. Though the data defined that low donor-type NK cell chimerism correlates with relapse, the association with GVHD was not reported.
In acute GVHD, reconstitution of donor NK cells is often delayed, and acute GVHD patients 2 months post-HSCT show lower numbers of CD56bright NK cells [168]. A prolonged increase in the number of CD56int NK cells has been observed in allo-HSCT patients with chronic GVHD [168]. As in murine pre-clinical studies, the presence of alloreactivity is required for human NK cell-mediated GVHD protection [182,183,184,185,186,187,188,189]. Most of these studies have focused on donor and/or recipient KIR receptor genotypes and haplotypes [182,183,184,185]. Lack of activating KIRs on donor NK cells (expected to be required for DC killing by NK cells) can increase GVHD risk [184]. When donor and recipient KIR genotypes are matched, the risk of chronic GVHD is diminished [183,185]. Next to matching donor and recipient KIR haplotypes, matching of donor KIR genotype with recipient HLA genotype appears to influence GVHD risk [186]. Donor→recipient combinations described worthy of avoidance in allo-HSCT are KIR2DL2→HLA-C2, KIR2DS2→HLA-C1, and KIR2DS1→HLA-C2 [186]. Recently, Hu et al. [190] noted a decrease in NKG2A+ NK cells in patients with GVHD as compared to those without GVHD after allo-HSCT. In vitro, NKG2A+ NK cells were able to suppress T cell IFN-γ production. Moreover, NKG2A+ NK cells were able to induce Tregs upon naive T cell co-culture [190]. In support of this, higher proportions of IFN-γ producing NK cells post-HSCT are associated with increased cumulative incidence of GVHD [191]. To summarize, it is important for OT induction that donor NK cells are “licensed” and that their inhibitory KIR are mismatched with the HLA ligands of recipients. Therefore, donor NK cell phenotype and recipient KIR and/or HLA status are important to consider when applying NK cells in allo-HSCT, not only for anti-tumor effects but also for tolerance induction [192]. Human ex vivo expanded NK cells have just completed phase I/II clinical trials. Whereas most of these have not shown successful GVHD reduction [193,194,195,196]. Lee and colleagues [197] did demonstrate GVHD reduction in patients given NK cell infusions. While others infused the NK cells after HSCT, Lee et al. did so prior to HSCT. Pre-clinical studies support that timing for NK cell infusion may be important for GVHD prevention [151,154]. Conversely, Shah et al. [198] reported aggravated acute GVHD upon NK cell infusion. Unlike the other trials, patients in this study received pre-activated NK cells. Production of pro-inflammatory cytokines from these pre-activated NK cells may in part explain why GVHD was worsened in this study. Though NK cells are established key effectors in anti-tumor immunotherapy, a better understanding of mechanisms employed by NK cells to direct immunotolerogenesis versus alloreactivity is needed to advance the application of NK cells in transplant tolerance induction.
4.2.2 Natural Killer T (NKT) Cells
Nomenclature and Phenotype. NKT cells (reviewed in [199]) share morphological and functional characteristics with both T cells and NK cells. Human NKT cells are mainly identified as CD3+TCRαβ+CD161+CD56+. Cytokines secreted by NKT cells can have powerful effects on αβ-T cell Th1/Th2 differentiation. NKT cells’ ability to rapidly respond without first having to differentiate into T effector cells places them at the front lines of defense against pathogens. NKT cells recognize antigen in the form of glycolipids presented by the MHC-I-like molecule CD1d on APCs; therefore, like NK cells, NKT cells are not restricted to MHC-I/II driven responses. NKT cells can be divided into 2 main subsets: Type I NKT cells, also known as invariant NKT (iNKT), and the less well understood type II NKT cells. iNKT cells express an invariant TCRα-chain (Vα14-Jα18 in mice and Vα24-Jα18 in humans) and can be further subdivided into CD4+CD8neg, CD4negCD8neg (termed “double negative” in mice and humans), and a rarer subset which is CD4negCD8+ [199].
Preclinical Data. Murine iNKT cells are particularly resistant to radiation-induced apoptosis due to their relatively high levels of anti-apoptotic factors including bcl-2 [200]. iNKT cells can rapidly secrete large amounts of cytokines including IL-4 upon activation [25,199]. We have demonstrated that preservation of iNKT cells in BM of recipient mice receiving MHC class I and class II-mismatched allo-HSCT was required for maintenance of bidirectional donor ←→ recipient immune tolerance and that iNKT cells allowed the maintenance of antitumor activity in the setting of HSCT for recipient-derived lymphoma [25,26]. We and others have shown, first in murine models and then in clinical HSCT, that this iNKT axis can be manipulated for transplant tolerance induction after allo-HSCT using reduced toxicity conditioning [25,26,201,202,203]. Others have since recapitulated that treatment of recipient mice by a variety of methods that enhance iNKT cell numbers or Th2 polarization can reduce GVHD morbidity and mortality [204,205,206,207,208]. Dominant innate immune mechanisms we have reported and/or others have recapitulated for iNKT cell-induced tolerance (Figure 2) include IL-4-dependent augmentation of PD-1 ligand-expressing recipient myeloid suppressor cells that induce contact-dependent expansion of donor thymically-derived Treg cells [25,26,27], costimulatory (CD40-CD40L) blockade of iNKT cells [207], direct inhibition of CD8+ T effector cells [25,209], and iNKT-associated generation of other subsets of tolerogenic CD8+ DCs [210]. In addition, there are other mechanisms not yet defined by which iNKT-derived Th2 cytokines may induce OT [200,201,202,209]. Adoptive transfers of iNKT cells with and without prior in vitro expansion have been successfully shown by us and other groups to reduce acute GVHD without impeding engraftment or anti-tumor activity [25,26,211,212,213,214,215].
Clinical Data. As important support for the relevance of iNKT cells to human immune tolerance, multiple groups in large-scale clinical studies have shown a strong association of patient overall survival and protection from GVHD on either iNKT cell graft content before HSCT [216] or early quantitative iNKT immune recovery after HSCT [217,218]. Peripheral blood stem cell CD4- iNKT cell dose was shown as the only graft parameter to predict significant acute GVHD, supposedly by selective suppression of T cell proliferation and IFN-γ secretion [216]. Just as in the pre-clinical setting, the same reduced toxicity pre-HSCT conditioning in patients increased IL-4 production by CD4+ T cells, elevated iNKT cell numbers, and reduced cumulative incidence of acute GVHD [19,219,220,221]. In our own data [222] and two separate recent publications [223,224], ex vivo expanded human iNKT cells were able to regulate T cell activation and proliferation while having direct in vitro and in vivo cytotoxicity against specific malignancies [225]. Thus cumulative current data indicate that iNKT cell-based immunotherapy is promising for reducing GVHD and allowing successful tolerance induction. Further investigation of tissue-specific iNKT interaction with other regulatory components of the immune system will likely pave the way for novel clinical protocols using human iNKT cells for tolerance induction after either HSCT or organ transplantation.
4.2.3 Gamma-Delta T (γδ-T) Cells
Nomenclature and Phenotype. γδ-T cells share many activating and inhibitory receptors with NK cells, and activation of these cells similarly depends on the net balance of signals from both types of receptors [226]. Like iNKT cells, γδ-T cells are CD3+ yet their TCR (in this case, TCRγδ) does not bind with antigen presented on conventional MHC molecules (reviewed in [227]). Instead, these cells recognize a broad range of ligands including phospholipids and small unprocessed peptides (reviewed in [228]). They can attack target cells directly through cytotoxic activity or indirectly through the activation of other immune cell subsets. γδT cell functional responses are induced upon the recognition of stress antigens, promoting cytokine production and regulating pathogen clearance [229]. In humans there are two major subsets of γδ-T cells identified by their Vδ chains. Vδ1-T cells are predominant in the thymus and peripheral tissues. Vδ2-T cells constitute the majority of circulating γδT cells [229]. Human γδ-T cells maintain a Vγ9 chain and mainly recognize phosphorylated non-peptide molecules that are metabolic intermediates of the isoprenoid biosynthesis pathway [229,230].
Preclinical Data. γδ-T cells are detected as early as 1 month post allo-HSCT in murine models using graft TCRαβ depletion [231]. The role of γδT cells in allo-tolerance is not entirely clear [229,230,231,232,233,234,235], particularly in mouse models. γδT cells have been reported to both reduce and increase GVHD depending upon the experimental model [231,232,233,234,235]. Drobyski et al. [232,236] showed that γδ-T cells do not induce GVHD. However, Blazar et al. [233] showed that the infusion of donor γδ-T cells did induce lethal GVHD in mice and Maeda and colleagues [234] observed similar effects of host γδ-T cells. In another study by Ellison et al. [235], elimination of these cells from donors significantly reduced GVHD and post-HSCT mortality.
Clinical Data. Partially mismatched or haploidentical allo-HSCT causes marked increases in the γδT cell pool and results in reduced GVHD in allo-HSCT for both non-malignant [231] and malignant disorders [237,238,239] (Figure 2), while allowing increased anti-tumor activity in allo-HSCT for malignancies [237,238,239]. Development of large-scale clinical methods for enriching, isolating, expanding, and therapeutically manipulating γδ‐T cells holds promise to ameliorate GVHD while maintaining anti-infectious immunity and enhancing long‐term survival after allo-HSCT [237,238,239].
4.3 Mesenchymal Stromal Cells (MSCs)
Nomenclature and Phenotype. MSCs are a fibroblast‐like, heterogeneous, non‐hematopoietic, pluripotent progenitor cell population with selfrenewal potential (reviewed in [240,241]). MSCs are plastic‐adherent and can be expanded in vitro. In humans, MSCs are characterized by the expression of CD73, CD90, and CD105 but absence of CD14, CD34, CD45 and HLA-DR. MSCs directly and indirectly suppress T cell reactivity (reviewed in [240,241]).
Preclinical Data. Infusion of allogeneic MSCs can induce immune tolerance to skin allografts [242]. MSCs have since been shown to rapidly migrate to sites of inflammation, including transplanted organs. MSCs exert immunomodulatory effects on a wide array of immune cells already known to play direct roles in transplantation tolerance, including T cells, B cells, NK cells, monocytes/macrophages, and DCs [242,243,244,245]. It is important to functionally distinguish MSCs (a stromal cell population derived from BM microenvironment) from MDSCs (immature myeloid hematopoietic cells of non-stromal origin) [243]. MSCs can arrest activated T cells in the G0/G1 phase and decrease their production of IFN‐γ and IL‐2 [243]. When exposed to inflammatory stimuli, as in the recently transplanted organ, MSCs display favorable immunomodulatory potential. This, combined with their unique migratory properties, make them strong theoretical candidates for site‐specific control of allograft inflammation [242,243,244,245]. Cho et al. [245] demonstrated that soluble factors in medium conditioned by human tonsil MSCs attenuates acute GVHD in a mouse model. Overall, MSC infusion in multiple transplant models results in a skew of the balance between regulatory and effector T cells towards a tolerogenic profile [243,244].
Clinical Data. Data support a role for MSCs in facilitating HSC engraftment and bidirectional tolerance, particularly in the setting of pediatric allo-HSCT [246,247,248]. In particular, donor-derived MSC co-transplantation following haploidentical allo-HSCT can facilitate engraftment with low rates of GVHD for severe aplastic anemia in children [248]. Ball et al. [249] showed that co-transplantation of ex vivo expanded MSCs may reduce the risk of graft failure in haploidentical HSCT. To interrogate the clinical utility of MSCs in reducing GVHD, a meta-analysis of randomized controlled trials (RCTs) was conducted [250]. In chronic GVHD patients with hematological malignancies undergoing allo-HSCT, umbilical cord-derived, high-dose, and late-infused MSCs prevented chronic GVHD (Figure 2), while bone marrow-derived, low-dose MSCs or MSCs co-infused with the graft appeared to be ineffective [250]. Though limited by the usual caveats of a meta-analysis, the study suggested that high-dose umbilical cord blood-derived MSC infusion should be explored in the RCT setting as a prophylactic strategy to reduce chronic GVHD after allo-HSCT. Three active clinical trials for HLA-matched and HLA-mismatched allo-HSCT in European, Israeli, and Korean consortia applying cord blood, bone marrow, or adipose tissue-derived MSCs to prevent or treat acute GVHD have yet to report their findings on safety and efficacy.
5. Future Directions
Medical and scientific advances since the first tissue transplants have greatly expanded the applications of both allo‐HSCT to cure hematopoietic and immune disorders and organ transplantation to prolong survival after organ failure. Nonetheless, relatively few significant strides have been made in the past 30‐40 years toward long‐range allograft survival and successful wean from pharmacological immunosuppression. Significant advances have recently been made in allo-HSCT by using reduced toxicity regimens that enhance recipient ←→ donor immune interactions and immune regulatory cell function. Other approaches remain limited by a requirement for long‐term immunosuppression, associated drug toxicity, and infectious and oncologic sequelae of chronic non-specific immune suppression. Our increased understanding of the regulatory cells and molecular pathways involved opens the opportunity for their exploitation to prevent and/or treat GVHD and graft rejection after allo-HSCT and, by extension, to prevent or treat organ allograft rejection. The theoretical advantages of the innate cellular immune regulatory approach are: 1) OT induction that obviates a need for ongoing immunosuppression and 2) alloantigen specificity, which reduces the risk of compromising immune responses to exogenous pathogens. To date, a number of key candidates have been identified in rigorous pre-clinical studies which appear to be translating well to human investigations, including iNKT cells, myeloid suppressor populations (MDSCs), and stromal cells (MSCs). Despite initial technical challenges relating to large‐scale expansion, these candidates are now undergoing clinical trials. Results from these and other studies over the next decade will likely greatly advance our understanding of the optimal regimens, the most appropriate cell sources (donor, host, third party), and any potential targeting of these cells required to improve long-range operational tolerance induction.
Author Contributions
Anouk A. J. Hamers, Sunil K. Joshi, and Asha B. Pillai performed the literature compilation, writing, editing, and proofreading.
Funding
This work was supported by grant #1R01HL133462 (NHLBI) (A.B.P.), the Batchelor Foundation for Pediatric Research (A.A.H., S.K.J., A.B.P.).
Competing Interests
The authors declare no financial or commercial conflict of interests.
References
- Murray JE. Human organ transplantation: Background and consequences. Science. 1992; 256: 1411-1416. [CrossRef]
- Gibson T, Medawar PB. The fate of skin homografts in man. J Anat. 1943; 77: 299-310.4.
- Brent L. A History of Transplantation Immunology. Academic Press; 1996. [CrossRef]
- Medawar PB. Tests by tissue culture methods on the nature of immunity to transplanted skin. Q J Microsc Sci. 1948; 89: 239-252. [CrossRef]
- Medawar PB. The cultivation of adult mammalian skin epithelium in vitro. Q J Microsc Sci. 1948; 89: 187-196. [CrossRef]
- Billingham RE, Brent L, Medawar PB. Quantitative studies on tissue transplantation immunity. II. The origin, strength and duration of actively and adoptively acquired immunity. Proc R Soc Lond B Biol Sci. 1954; 143: 58-80. [CrossRef]
- Ruiz P, Maldonado P, Hidalgo Y, Gleisner A, Sauma D, Silva C, et al. Transplant tolerance: New insights and strategies for long-term allograft acceptance. Clin Dev Immunol. 2013; 2013: 210506. [CrossRef]
- Thomas ED, Storb R, Clift RA, Fefer A, Johnson L, Neiman PE, et al. Bone-marrow transplantation (second of two parts). N Engl J Med. 1975; 292: 895-902. [CrossRef]
- Thomas E, Storb R, Clift RA, Fefer A, Johnson FL, Neiman PE, et al. Bone-marrow transplantation (first of two parts). N Engl J Med. 1975; 292: 832-843. [CrossRef]
- Appelbaum FR. Hematopoietic-cell transplantation at 50. N Engl J Med. 2007; 357: 14721475. [CrossRef]
- Thiel G, Harder F, Lörtscher R, Brünisholz M, Landmann J, Brunner F, et al. Cyclosporin a used alone or in combination with low-dose steroids in cadaveric renal transplantation. Klin Wochenschr. 1983; 61: 991-1000. [CrossRef]
- Tedesco D, Haragsim L. Cyclosporine: A review. J Transplant. 2012; 2012: 230386. [CrossRef]
- Filippini P, Rutella S. Recent advances on cellular therapies and immune modulators for graft-versus-host disease. Expert Rev Clin Immunol. 2014; 10: 1357-1374. [CrossRef]
- Fehr T, Sykes M. Tolerance induction in clinical transplantation. Transpl Immunol. 2004; 13: 117-130. [CrossRef]
- Xing Y, Hogquist KA. T-cell tolerance: Central and peripheral. Cold Spring Harb Perspect Biol. 2012; 4. doi:10.1101/cshperspect.a006957. [CrossRef]
- Mueller DL. Mechanisms maintaining peripheral tolerance. Nat Immunol. 2010; 11: 21-27. [CrossRef]
- Fuchs EJ. Transplantation tolerance: From theory to clinic. Immunol Rev. 2014; 258: 64-79. [CrossRef]
- Kawai T, Leventhal J, Wood K, Strober S. Summary of the third international workshop on clinical tolerance. Am J Transplant. 2018. doi:10.1111/ajt.15086. [CrossRef]
- Lowsky R, Takahashi T, Liu YP, Dejbakhsh-Jones S, Grumet FC, Shizuru JA, et al. Protective conditioning for acute graft-versus-host disease. N Engl J Med. 2005; 353: 1321-1331. [CrossRef]
- Millan MT, Shizuru JA, Hoffmann P, Dejbakhsh-Jones S, Scandling JD, Grumet FC, et al. Mixed chimerism and immunosuppressive drug withdrawal after HLA-mismatched kidney and hematopoietic progenitor transplantation. Transplantation. 2002; 73: 1386-1391. [CrossRef]
- Sykes M, Sachs DH. Mixed chimerism. Philos Trans R Soc Lond B Biol Sci. 2001; 356: 707-726. [CrossRef]
- Sachs DH, Sykes M, Kawai T, Cosimi AB. Immuno-intervention for the induction of transplantation tolerance through mixed chimerism. Semin Immunol. 2011; 23: 165-173. [CrossRef]
- Sachs DH, Kawai T, Sykes M. Induction of tolerance through mixed chimerism. Cold Spring Harb Perspect Med. 2014; 4: a015529. [CrossRef]
- Zuber J, Sykes M. Mechanisms of mixed chimerism-based transplant tolerance. Trends Immunol. 2017; 38: 829-843. [CrossRef]
- Pillai AB, George TI, Dutt S, Strober S. Host natural killer T cells induce an interleukin-4dependent expansion of donor CD4+CD25+Foxp3+ T regulatory cells that protects against graft-versus-host disease. Blood. 2009; 113: 4458-4467. [CrossRef]
- Pillai AB, George TI, Dutt S, Teo P, Strober S. Host NKT cells can prevent graft-versus-host disease and permit graft antitumor activity after bone marrow transplantation. J Immunol. 2007; 178: 6242-6251. [CrossRef]
- E S, Seth A, Vogel P, Sommers M, Ong T, Pillai AB. Bidirectional immune tolerance in nonmyeloablative MHC-mismatched BMT for murine β-thalassemia. Blood. 2017; 129: 30173030. [CrossRef]
- van der Merwe M, Abdelsamed HA, Seth A, Ong T, Vogel P, Pillai AB. Recipient myeloidderived immunomodulatory cells induce PD-1 ligand-dependent donor CD4+Foxp3+ regulatory T cell proliferation and donor-recipient immune tolerance after murine nonmyeloablative bone marrow transplantation. J Immunol. 2013; 191: 5764-5776. [CrossRef]
- Guan Q, Blankstein AR, Anjos K, Synova O, Tulloch M, Giftakis A, et al. Functional myeloidderived suppressor cell subsets recover rapidly after allogeneic hematopoietic stem/progenitor cell transplantation. Biol Blood Marrow Transplant. 2015; 21: 1205-1214. [CrossRef]
- Yin J, Wang C, Huang M, Mao X, Zhou J, Zhang Y. Circulating CD14 HLA-DR-/lowmyeloidderived suppressor cells in leukemia patients with allogeneic hematopoietic stem cell transplantation: Novel clinical potential strategies for the prevention and cellular therapy of graft-versus-host disease. Cancer Med. 2016; 5: 1654-1669. [CrossRef]
- Tcyganov E, Mastio J, Chen E, Gabrilovich DI. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol. 2018; 51: 76-82. [CrossRef]
- Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: Immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. 2018; 200: 422-431. [CrossRef]
- Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017; 5: 3-8. [CrossRef]
- Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010; 22: 238-244. [CrossRef]
- Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016; 7: 12150. [CrossRef]
- Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity. 2016; 44: 303-315. [CrossRef]
- Pillay J, Tak T, Kamp VM, Koenderman L. Immune suppression by neutrophils and granulocytic myeloid-derived suppressor cells: Similarities and differences. Cell Mol Life Sci. 2013; 70: 3813-3827. [CrossRef]
- Aarts CEM, Kuijpers TW. Neutrophils as myeloid-derived suppressor cells. Eur J Clin Invest. 2018; 48: e12989. [CrossRef]
- Bergenfelz C, Larsson A-M, von Stedingk K, Gruvberger-Saal S, Aaltonen K, Jansson S, et al. Systemic monocytic-MDSCs are generated from monocytes and correlate with disease progression in breast cancer patients. PLoS One. 2015; 10: e0127028. [CrossRef]
- Mao Y, Poschke I, Wennerberg E, Pico de Coaña Y, Egyhazi Brage S, Schultz I, et al. Melanoma-educated CD14+ cells acquire a myeloid-derived suppressor cell phenotype through COX-2-dependent mechanisms. Cancer Res. 2013; 73: 3877-3887. [CrossRef]
- Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. 2013; 123: 4464-4478. [CrossRef]
- Gato-Cañas M, Martinez de Morentin X, Blanco-Luquin I, Fernandez-Irigoyen J, Zudaire I, Liechtenstein T, et al. A core of kinase-regulated interactomes defines the neoplastic MDSC lineage. Oncotarget. 2015; 6: 27160-27175. [CrossRef]
- Luyckx A, Schouppe E, Rutgeerts O, Lenaerts C, Koks C, Fevery S, et al. Subset characterization of myeloid-derived suppressor cells arising during induction of BM chimerism in mice. Bone Marrow Transplant. 2012; 47: 985-992. [CrossRef]
- Wang D, Yu Y, Haarberg K, Fu J, Kaosaard K, Nagaraj S, et al. Dynamic change and impact of myeloid-derived suppressor cells in allogeneic bone marrow transplantation in mice. Biol Blood Marrow Transplant. 2013; 19: 692-702. [CrossRef]
- Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010; 116: 5738-5747. [CrossRef]
- Messmann JJ, Reisser T, Leithäuser F, Lutz MB, Debatin K-M, Strauss G. In vitro-generated MDSCs prevent murine GVHD by inducing type 2 T cells without disabling antitumor cytotoxicity. Blood. 2015; 126: 1138-1148. [CrossRef]
- MacDonald KPA, Rowe V, Clouston AD, Welply JK, Kuns RD, Ferrara JLM, et al. Cytokine expanded myeloid precursors function as regulatory antigen-presenting cells and promote tolerance through IL-10-producing regulatory T cells. J Immunol. 2005; 174: 1841-1850. [CrossRef]
- Lim J-Y, Ryu D-B, Park M-Y, Lee S-E, Park G, Kim T-G, et al. Ex vivo generated human cord blood myeloid-derived suppressor cells attenuate murine chronic graft-versus-host diseases. Biol Blood Marrow Transplant. 2018; 24: 2381-2396. [CrossRef]
- Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med. 2002; 196: 389-399. [CrossRef]
- Colonna L, Sega EI, Negrin RS. Natural and expanded CD4(+)CD25(+) regulatory T cells in bone marrow transplantation. Biol Blood Marrow Transplant. 2011; 17: S58-S62. [CrossRef]
- Park M-J, Lee S-H, Kim E-K, Lee E-J, Baek J-A, Park S-H, et al. Interleukin-10 produced by myeloid-derived suppressor cells is critical for the induction of Tregs and attenuation of rheumatoid inflammation in mice. Sci Rep. 2018; 8: 3753. [CrossRef]
- Lee CR, Kwak Y, Yang T, Han JH, Park SH, Ye MB, et al. Myeloid-derived suppressor cells are controlled by regulatory T cells via TGF-β during murine colitis. Cell Rep. 2016; 17: 32193232. [CrossRef]
- Kang X, Zhang X, Liu Z, Xu H, Wang T, He L, et al. Granulocytic myeloid-derived suppressor cells maintain feto-maternal tolerance by inducing Foxp3 expression in CD4 CD25−T cells by activation of the TGF-β/β-catenin pathway. Mol Hum Reprod. 2016; 22: 499-511. [CrossRef]
- Zhou Z, French DL, Ma G, Eisenstein S, Chen Y, Divino CM, et al. Development and function of myeloid-derived suppressor cells generated from mouse embryonic and hematopoietic stem cells. Stem Cells. 2010. doi:10.1002/stem.301. [CrossRef]
- Matta BM, Reichenbach DK, Zhang X, Mathews L, Koehn BH, Dwyer GK, et al. Peri-alloHCT IL33 administration expands recipient T-regulatory cells that protect mice against acute GVHD. Blood. 2016; 128: 427-439. [CrossRef]
- Drujont L, Carretero-Iglesia L, Bouchet-Delbos L, Beriou G, Merieau E, Hill M, et al. Evaluation of the therapeutic potential of bone marrow-derived myeloid suppressor cell (MDSC) adoptive transfer in mouse models of autoimmunity and allograft rejection. PLoS One. 2014; 9: e100013. [CrossRef]
- Mougiakakos D, Jitschin R, von Bahr L, Poschke I, Gary R, Sundberg B, et al. Immunosuppressive CD14+HLA-DRlow/neg IDO+ myeloid cells in patients following allogeneic hematopoietic stem cell transplantation. Leukemia. 2013; 27: 377-388. [CrossRef]
- Testa U, Martucci R, Rutella S, Scambia G, Sica S, Benedetti Panici P, et al. Autologous stem cell transplantation: release of early and late acting growth factors relates with hematopoietic ablation and recovery. Blood. 1994; 84: 3532-3539. [CrossRef]
- Janikashvili N, Trad M, Gautheron A, Samson M, Lamarthée B, Bonnefoy F, et al. Human monocyte-derived suppressor cells control graft-versus-host disease by inducing regulatory forkhead box protein 3-positive CD8+ T lymphocytes. J Allergy Clin Immunol. 2015; 135: 1614-1624.e4. [CrossRef]
- Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. 2010; 185: 2273-2284. [CrossRef]
- Koehn BH, Apostolova P, Haverkamp JM, Miller JS, McCullar V, Tolar J, et al. GVHDassociated, inflammasome-mediated loss of function in adoptively transferred myeloidderived suppressor cells. Blood. 2015; 126: 1621-1628. [CrossRef]
- Tacke RS, Lee H-C, Goh C, Courtney J, Polyak SJ, Rosen HR, et al. Myeloid suppressor cells induced by hepatitis C virus suppress T-cell responses through the production of reactive oxygen species. Hepatology. 2012; 55: 343-353. [CrossRef]
- Hutchinson JA, Riquelme P, Geissler EK. Human regulatory macrophages as a cell-based medicinal product. Curr Opin Organ Transplant. 2012; 17: 48-54. [CrossRef]
- Lavin Y, Merad M. Macrophages: gatekeepers of tissue integrity. Cancer Immunol Res. 2013; 1: 201-209. [CrossRef]
- Alegre M-L, Goldstein DR, Chong AS. Toll-like receptor signaling in transplantation. Curr Opin Organ Transplant. 2008; 13: 358-365. [CrossRef]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004; 25: 677686. [CrossRef]
- Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014; 6: 13. [CrossRef]
- Broichhausen C, Riquelme P, Geissler EK, Hutchinson JA. Regulatory macrophages as therapeutic targets and therapeutic agents in solid organ transplantation. Curr Opin Organ Transplant. 2012; 17: 332-342. [CrossRef]
- Salehi S, Reed EF. The divergent roles of macrophages in solid organ transplantation. Curr Opin Organ Transplant. 2015; 20: 446-453. [CrossRef]
- Zhao HY, Lyu ZS, Duan CW, Song Y, Han TT, Mo XD, et al. An unbalanced monocyte macrophage polarization in the bone marrow microenvironment of patients with poor graft function after allogeneic haematopoietic stem cell transplantation. Br J Haematol. 2018; 182: 679-692. [CrossRef]
- Xu H, Yan J, Zhu Z, Hussain LR, Huang Y, Ding C, et al. A critical role for the TLR4/TRIF pathway in allogeneic hematopoietic cell rejection by innate immune cells. Cell Transplant. 2013; 22: 2367-2380. [CrossRef]
- Thompson HL, van Rooijen N, McLelland BT, Manilay JO. F4/80 host macrophages are a barrier to murine embryonic stem cell-derived hematopoietic progenitor engraftment in vivo. J Immunol Res. 2016; 2016: 1-12. [CrossRef]
- Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JL. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood. 1997; 90: 3204-3213. [CrossRef]
- Imado T, Iwasaki T, Kitano S, Satake A, Kuroiwa T, Tsunemi S, et al. The protective role of host Toll-like receptor-4 in acute graft-versus-host disease. Transplantation. 2010; 90: 10631070. [CrossRef]
- Hashimoto D, Chow A, Greter M, Saenger Y, Kwan W-H, Leboeuf M, et al. Pretransplant CSF1 therapy expands recipient macrophages and ameliorates GVHD after allogeneic hematopoietic cell transplantation. J Exp Med. 2011; 208: 1069-1082. [CrossRef]
- Yamakawa T, Ohigashi H, Hashimoto D, Hayase E, Takahashi S, Miyazaki M, et al. Vitamin Acoupled liposomes containing siRNA against HSP47 ameliorate skin fibrosis in chronic graftversus-host disease. Blood. 2018; 131: 1476-1485. [CrossRef]
- Du J, Paz K, Flynn R, Vulic A, Robinson TM, Lineburg KE, et al. Pirfenidone ameliorates murine chronic GVHD through inhibition of macrophage infiltration and TGF-β production. Blood. 2017; 129: 2570-2580. [CrossRef]
- Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M, et al. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J Biol Chem. 2012; 287: 11629-11641. [CrossRef]
- Mukai S, Ogawa Y, Urano F, Kudo-Saito C, Kawakami Y, Tsubota K. Novel treatment of chronic graft-versus-host disease in mice using the ER stress reducer 4-Phenylbutyric acid. Sci Rep. 2017; 7: 41939. [CrossRef]
- Kawashima N, Terakura S, Nishiwaki S, Koyama D, Ozawa Y, Ito M, et al. Increase of bone marrow macrophages and CD8 T lymphocytes predict graft failure after allogeneic bone marrow or cord blood transplantation. Bone Marrow Transplant. 2017; 52: 1164-1170. [CrossRef]
- Hyvärinen K, Ritari J, Koskela S, Niittyvuopio R, Nihtinen A, Volin L, et al. Genetic polymorphism related to monocyte-macrophage function is associated with graft-versushost disease. Sci Rep. 2017; 7: 15666. [CrossRef]
- Nestel FP, Price KS, Seemayer TA, Lapp WS. Macrophage priming and lipopolysaccharidetriggered release of tumor necrosis factor alpha during graft-versus-host disease. J Exp Med. 1992; 175: 405-413. [CrossRef]
- Hueso T, Coiteux V, Curt MJC, Labreuche J, Jouault T, Yakoub-Agha I, et al. Citrulline and monocyte-derived macrophage reactivity before conditioning predict acute graft-versushost disease. Biol Blood Marrow Transplant. 2017; 23: 913-921. [CrossRef]
- Haniffa M, Ginhoux F, Wang X-N, Bigley V, Abel M, Dimmick I, et al. Differential rates of replacement of human dermal dendritic cells and macrophages during hematopoietic stem cell transplantation. J Exp Med. 2009; 206: 371-385. [CrossRef]
- Inamoto Y, Martin PJ, Paczesny S, Tabellini L, Momin AA, Mumaw CL, et al. Association of plasma CD163 concentration with de novo-onset chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2017; 23: 1250-1256. [CrossRef]
- Buechler C, Ritter M, Orsó E, Langmann T, Klucken J, Schmitz G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J Leukoc Biol. 2000; 67: 97-103. [CrossRef]
- Hutchinson JA, Riquelme P, Sawitzki B, Tomiuk S, Miqueu P, Zuhayra M, et al. Cutting edge: Immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J Immunol. 2011; 187: 2072-2078. [CrossRef]
- van Kooten C, Lombardi G, Gelderman KA, Sagoo P, Buckland M, Lechler R, et al. Dendritic cells as a tool to induce transplantation tolerance: obstacles and opportunities. Transplantation. 2011; 91: 2-7. [CrossRef]
- Obregon C, Kumar R, Pascual MA, Vassalli G, Golshayan D. Update on dendritic cell-induced immunological and cclinical tolerance. Front Immunol. 2017; 8: 1514. [CrossRef]
- Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013; 31: 563-604. [CrossRef]
- Schlitzer A, Zhang W, Song M, Ma X. Recent advances in understanding dendritic cell development, classification, and phenotype. F1000Res. 2018; 7. doi:10.12688/f1000research.14793.1. [CrossRef]
- Thompson AG, O’Sullivan BJ, Beamish H, Thomas R. T cells signaled by NF-kappa B- dendritic cells are sensitized not anergic to subsequent activation. J Immunol. 2004; 173: 1671-1680. [CrossRef]
- Sayegh MH, Turka LA. The role of T-cell costimulatory activation pathways in transplant rejection. N Engl J Med. 1998; 338: 1813-1821. [CrossRef]
- Benichou G, Thomson AW. Direct versus indirect allorecognition pathways: On the right track. Am J Transplant. 2009; 9: 655-656. [CrossRef]
- Liu Z. Contribution of direct and indirect recognition pathways to T cell alloreactivity. J Exp Med. 1993; 177: 1643-1650. [CrossRef]
- Ciubotariu R, Liu Z, Colovai AI, Ho E, Itescu S, Ravalli S, et al. Persistent allopeptide reactivity and epitope spreading in chronic rejection of organ allografts. J Clin Invest. 1998; 101: 398405. [CrossRef]
- Marín E, Cuturi MC, Moreau A. Tolerogenic dendritic cells in solid organ transplantation: Where do we stand? Front Immunol. 2018; 9: 274. [CrossRef]
- Merad M, Hoffmann P, Ranheim E, Slaymaker S, Manz MG, Lira SA, et al. Depletion of host Langerhans cells before transplantation of donor alloreactive T cells prevents skin graftversus-host disease. Nat Med. 2004; 10: 510-517. [CrossRef]
- Li H, Kaplan DH, Matte-Martone C, Tan HS, Venkatesan S, Johnson K, et al. Langerhans cells are not required for graft-versus-host disease. Blood. 2010; 117: 697-707. [CrossRef]
- Mengwasser J, Babes L, Cordes S, Mertlitz S, Riesner K, Shi Y, et al. Cathepsin E deficiency ameliorates graft-versus-host disease and modifies dendritic cell motility. Front Immunol. 2017; 8: 203. [CrossRef]
- Chen S, Smith BAH, Iype J, Prestipino A, Pfeifer D, Grundmann S, et al. MicroRNA-155deficient dendritic cells cause less severe GVHD through reduced migration and defective inflammasome activation. Blood. 2015; 126: 103-112. [CrossRef]
- Wilhelm K, Ganesan J, Müller T, Dürr C, Grimm M, Beilhack A, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med. 2010; 16: 1434-1438. [CrossRef]
- Zhong X, Zhu F, Qiao J, Zhao K, Zhu S, Zeng L, et al. The impact of P2X7 receptor antagonist, brilliant blue G on graft-versus-host disease in mice after allogeneic hematopoietic stem cell transplantation. Cell Immunol. 2016; 310: 71-77. [CrossRef]
- Geraghty NJ, Watson D, Sluyter R. Long-term treatment with the P2X7 receptor antagonist Brilliant Blue G reduces liver inflammation in a humanized mouse model of graft-versus-host disease. Cell Immunol. 2018. doi:10.1016/j.cellimm.2018.12.001. [CrossRef]
- Leveque-El Mouttie L, Koyama M, Le Texier L, Markey KA, Cheong M, Kuns RD, et al. Corruption of dendritic cell antigen presentation during acute GVHD leads to regulatory Tcell failure and chronic GVHD. Blood. 2016; 128: 794-804. [CrossRef]
- Iberg CA, Jones A, Hawiger D. Dendritic cells as inducers of peripheral tolerance. Trends Immunol. 2017; 38: 793-804. [CrossRef]
- Hasegawa H, Matsumoto T. Mechanisms of tolerance induction by dendritic cells in vivo. Front Immunol. 2018; 9. doi:10.3389/fimmu.2018.00350. [CrossRef]
- Dalmasso AP, Martinez C, Sjodin K, Good RA. Studies on the role of the thymus in immunobiology; reconstitution of immunologic capacity in mice thymectomized at birth. J Exp Med. 1963; 118: 1089-1109. [CrossRef]
- Stenger EO, Rosborough BR, Mathews LR, Ma H, Mapara MY, Thomson AW, et al. IL-12hi rapamycin-conditioned dendritic cells mediate IFN-γ-dependent apoptosis of alloreactive CD4+ T cells in vitro and reduce lethal graft-versus-host disease. Biol Blood Marrow Transplant. 2014; 20: 192-201. [CrossRef]
- Comi M, Amodio G, Gregori S. Interleukin-10-producing DC-10 is a unique tool to promote tolerance via antigen-specific T regulatory type 1 cells. Front Immunol. 2018; 9. doi:10.3389/fimmu.2018.00682. [CrossRef]
- Fugier-Vivier IJ, Rezzoug F, Huang Y, Graul-Layman AJ, Schanie CL, Xu H, et al. Plasmacytoid precursor dendritic cells facilitate allogeneic hematopoietic stem cell engraftment. J Exp Med. 2005; 201: 373-383. [CrossRef]
- Huang Y, Bozulic LD, Miller T, Xu H, -R. Hussain L, Ildstad ST. CD8 plasmacytoid precursor DCs induce antigen-specific regulatory T cells that enhance HSC engraftment in vivo. Blood. 2010; 117: 2494-2505. [CrossRef]
- Taylor KN, Shinde-Patil VR, Cohick E, Colson YL. Induction of FoxP3+CD4+25+ regulatory T cells following hemopoietic stem cell transplantation: Role of bone marrow-derived facilitating cells. J Immunol. 2007; 179: 2153-2162. [CrossRef]
- Li H, Demetris AJ, McNiff J, Matte-Martone C, Tan HS, Rothstein DM, et al. Profound depletion of host conventional dendritic cells, plasmacytoid dendritic cells, and B cells does not prevent graft-versus-host disease induction. J Immunol. 2012; 188: 3804-3811. [CrossRef]
- Banovic T, Markey KA, Kuns RD, Olver SD, Raffelt NC, Don AL, et al. Graft-versus-host disease prevents the maturation of plasmacytoid dendritic cells. J Immunol. 2009; 182: 912-920. [CrossRef]
- Li J-M, Southerland LT, Lu Y, Darlak KA, Giver CR, McMillin DW, et al. Activation, immune polarization, and graft-versus-leukemia activity of donor T cells are regulated by specific subsets of donor bone marrow antigen-presenting cells in allogeneic hemopoietic stem cell transplantation. J Immunol. 2009; 183: 7799-7809. [CrossRef]
- Klangsinsirikul P, Carter GI, Byrne JL, Hale G, Russell NH. Campath-1G causes rapid depletion of circulating host dendritic cells (DCs) before allogeneic transplantation but does not delay donor DC reconstitution. Blood. 2002; 99: 2586-2591. [CrossRef]
- Auffermann-Gretzinger S, Lossos IS, Vayntrub TA, Leong W, Grumet FC, Blume KG, et al. Rapid establishment of dendritic cell chimerism in allogeneic hematopoietic cell transplant recipients. Blood. 2002; 99: 1442-1448. [CrossRef]
- Perreault C, Pelletier M, Belanger R, Boileau J, Bonny Y, David M, et al. Persistence of host Langerhans cells following allogeneic bone marrow transplantation: Possible relationship with acute graft-versus-host disease. Br J Haematol. 1985; 60: 253-260. [CrossRef]
- Horváth R, Budinský V, Kayserová J, Kalina T, Formánková R, Starý J, et al. Kinetics of dendritic cells reconstitution and costimulatory molecules expression after myeloablative allogeneic haematopoetic stem cell transplantation: Implications for the development of acute graft-versus host disease. Clin Immunol. 2009; 131: 60-69. [CrossRef]
- Vakkila J, Thomson AW, Hovi L, Vettenranta K, Saarinen-Pihkala UM. Circulating dendritic cell subset levels after allogeneic stem cell transplantation in children correlate with time post transplant and severity of acute graft-versus-host disease. Bone Marrow Transplant. 2005; 35: 501-507. [CrossRef]
- Chan GW, Gorgun G, Miller KB, Foss FM. Persistence of host dendritic cells after transplantation is associated with graft-versus-host disease. Biol Blood Marrow Transplant. 2003; 9: 170-176. [CrossRef]
- Clark FJ, Freeman L, Dzionek A, Schmitz J, McMullan D, Simpson P, et al. Origin and subset distribution of peripheral blood dendritic cells in patients with chronic graft-versus-host disease. Transplantation. 2003; 75: 221-225. [CrossRef]
- Rajasekar R, Lakshmi KM, George B, Viswabandya A, Thirugnanam R, Abraham A, et al. Dendritic cell count in the graft predicts relapse in patients with hematologic malignancies undergoing an HLA-matched related allogeneic peripheral blood stem cell transplant. Biol Blood Marrow Transplant. 2010; 16: 854-860. [CrossRef]
- Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001; 193: 233-238. [CrossRef]
- Dhodapkar MV, Steinman RM. Antigen-bearing immature dendritic cells induce peptidespecific CD8(+) regulatory T cells in vivo in humans. Blood. 2002; 100: 174-177. [CrossRef]
- Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. 2011; 34: 2026-2032. [CrossRef]
- Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med. 2015; 7: 290ra87. [CrossRef]
- Jauregui-Amezaga A, Cabezón R, Ramírez-Morros A, España C, Rimola J, Bru C, et al. Intraperitoneal administration of autologous tolerogenic dendritic cells for refractory crohn’s disease: A phase I study. J Crohns Colitis. 2015; 9: 1071-1078. [CrossRef]
- Rosborough BR, Raïch-Regué D, Turnquist HR, Thomson AW. Regulatory myeloid cells in transplantation. Transplantation. 2014; 97: 367-379. [CrossRef]
- Steinbrink K, Wölfl M, Jonuleit H, Knop J, Enk AH. Induction of tolerance by IL-10-treated dendritic cells. J Immunol. 1997; 159: 4772-4780.
- Boks MA, Kager-Groenland JR, Haasjes MSP, Zwaginga JJ, van Ham SM, ten Brinke A. IL-10generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction--a comparative study of human clinical-applicable DC. Clin Immunol. 2012; 142: 332-342. [CrossRef]
- Fogel-Petrovic M, Long JA, Misso NL, Foster PS, Bhoola KD, Thompson PJ. Physiological concentrations of transforming growth factor beta1 selectively inhibit human dendritic cell function. Int Immunopharmacol. 2007; 7: 1924-1933. [CrossRef]
- Fedoric B, Krishnan R. Rapamycin downregulates the inhibitory receptors ILT2, ILT3, ILT4 on human dendritic cells and yet induces T cell hyporesponsiveness independent of FoxP3 induction. Immunol Lett. 2008; 120: 49-56. [CrossRef]
- Naranjo-Gómez M, Raïch-Regué D, Oñate C, Grau-López L, Ramo-Tello C, Pujol-Borrell R, et al. Comparative study of clinical grade human tolerogenic dendritic cells. J Transl Med. 2011; 9: 89. [CrossRef]
- de Jong EC, Vieira PL, Kalinski P, Kapsenberg ML. Corticosteroids inhibit the production of inflammatory mediators in immature monocyte-derived DC and induce the development of tolerogenic DC3. J Leukoc Biol. 1999; 66: 201-204. [CrossRef]
- Xia C-Q, Peng R, Beato F, Clare-Salzler MJ. Dexamethasone induces IL-10-producing monocyte-derived dendritic cells with durable immaturity. Scand J Immunol. 2005; 62: 45-54. [CrossRef]
- Cabezón R, Carrera-Silva EA, Flórez-Grau G, Errasti AE, Calderón-Gómez E, Lozano JJ, et al. MERTK as negative regulator of human T cell activation. J Leukoc Biol. 2015; 97: 751-760. [CrossRef]
- Penna G, Adorini L. 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol. 2000; 164: 2405-2411. [CrossRef]
- Zahorchak AF, Macedo C, Hamm DE, Butterfield LH, Metes DM, Thomson AW. High PDL1/CD86 MFI ratio and IL-10 secretion characterize human regulatory dendritic cells generated for clinical testing in organ transplantation. Cell Immunol. 2018; 323: 9-18. [CrossRef]
- Carotta S. Targeting NK cells for anticancer immunotherapy: Clinical and preclinical approaches. Front Immunol. 2016; 7: 152. [CrossRef]
- Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol. 2006; 176: 1517-1524. [CrossRef]
- Handgretinger R, Lang P, André MC. Exploitation of natural killer cells for the treatment of acute leukemia. Blood. 2016; 127: 3341-3349. [CrossRef]
- Yokoyama WM, Kim S. Licensing of natural killer cells by self-major histocompatibility complex class I. Immunol Rev. 2006; 214: 143-154. [CrossRef]
- Gasser S, Raulet DH. Activation and self-tolerance of natural killer cells. Immunol Rev. 2006; 214: 130-142. [CrossRef]
- Anfossi N, André P, Guia S, Falk CS, Roetynck S, Stewart CA, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006; 25: 331-342. [CrossRef]
- Sungur CM, Tang-Feldman YJ, Zamora AE, Alvarez M, Pomeroy C, Murphy WJ. Murine NKcell licensing is reflective of donor MHC-I following allogeneic hematopoietic stem cell transplantation in murine cytomegalovirus responses. Blood. 2013; 122: 1518-1521. [CrossRef]
- Farnsworth A, Wotherspoon JS, Dorsch SE. Postirradiation recovery of lymphoid cells in the rat. Transplantation. 1988; 46: 418-425. [CrossRef]
- Glineur S, Antoine-Moussiaux N, Michaux C, Desmecht D. Immune depression of the SJL/J mouse, a radioresistant and immunologically atypical inbred strain. Immunobiology. 2011; 216: 213-217. [CrossRef]
- Sun K, Alvarez M, Ames E, Barao I, Chen M, Longo DL, et al. Mouse NK cell-mediated rejection of bone marrow allografts exhibits patterns consistent with Ly49 subset licensing. Blood. 2012; 119: 1590-1598. [CrossRef]
- Asai O, Longo DL, Tian ZG, Hornung RL, Taub DD, Ruscetti FW, et al. Suppression of graftversus-host disease and amplification of graft-versus-tumor effects by activated natural killer cells after allogeneic bone marrow transplantation. J Clin Invest. 1998; 101: 1835-1842. [CrossRef]
- Lundqvist A, McCoy JP, Samsel L, Childs R. Reduction of GVHD and enhanced antitumor effects after adoptive infusion of alloreactive Ly49-mismatched NK cells from MHC-matched donors. Blood. 2007; 109: 3603-3606. [CrossRef]
- Chen G, Wu D, Wang Y, Cen J, Feng Y, Sun A, et al. Expanded donor natural killer cell and IL-2, IL-15 treatment efficacy in allogeneic hematopoietic stem cell transplantation. Eur J Haematol. 2008; 81: 226-235. [CrossRef]
- Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood. 2010; 115: 4293-4301. [CrossRef]
- Hüber CM, Doisne J-M, Colucci F. IL-12/15/18-preactivated NK cells suppress GvHD in a mouse model of mismatched hematopoietic cell transplantation. Eur J Immunol. 2015; 45: 1727-1735. [CrossRef]
- Ruggeri L. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002; 295: 2097-2100. [CrossRef]
- Cao D, Hu L, Wang Y, Wang L, Zheng W, Ma W. Suppression of graft-versus-host disease after adoptive infusion of alloreactive NK cells induced by silencing Ly49C gene in mice. Transpl Immunol. 2009; 20: 243-248. [CrossRef]
- Chow T, Whiteley J, Li M, Rogers IM. The transfer of host MHC class I protein protects donor cells from NK cell and macrophage-mediated rejection during hematopoietic stem cell transplantation and engraftment in mice. Stem Cells. 2013; 31: 2242-2252. [CrossRef]
- Meinhardt K, Kroeger I, Bauer R, Ganss F, Ovsiy I, Rothamer J, et al. Identification and characterization of the specific murine NK cell subset supporting graft--leukemia- and reducing graft--host-effects. Oncoimmunology. 2015; 4: e981483. [CrossRef]
- Ghadially H, Ohana M, Elboim M, Gazit R, Gur C, Nagler A, et al. NK cell receptor NKp46 regulates graft-versus-host disease. Cell Rep. 2014; 7: 1809-1814. [CrossRef]
- Yu G, Xu X, Vu MD, Kilpatrick ED, Li XC. NK cells promote transplant tolerance by killing donor antigen-presenting cells. J Exp Med. 2006; 203: 1851-1858. [CrossRef]
- Laffont S, Seillet C, Ortaldo J, Coudert JD, Guéry J-C. Natural killer cells recruited into lymph nodes inhibit alloreactive T-cell activation through perforin-mediated killing of donor allogeneic dendritic cells. Blood. 2008; 112: 661-671. [CrossRef]
- Rabinovich BA, Li J, Shannon J, Hurren R, Chalupny J, Cosman D, et al. Activated, but not resting, T cells can be recognized and killed by syngeneic NK cells. J Immunol. 2003; 170: 3572-3576. [CrossRef]
- Ellison CA, HayGlass KT, Fischer JM, Rector ES, MacDonald GC, Gartner JG. Depletion of natural killer cells from the graft reduces interferon-gamma levels and lipopolysaccharideinduced tumor necrosis factor-alpha release in F1 hybrid mice with acute graft-versus-host disease. Transplantation. 1998; 66: 284-294. [CrossRef]
- Xun C, Brown SA, Darrell Jennings C, Jean Henslee-Downey P, Thompson JS. Acute graftversus-host-like disease induced by transplantation of human activated natural killer cells into SCID mice. Transplantation. 1993; 56: 409-416. [CrossRef]
- Xun CQ, Thompson JS, Jennings CD, Brown SA. The effect of human IL-2-activated natural killer and T cells on graft-versus-host disease and graft-versus-leukemia in SCID mice bearing human leukemic cells. Transplantation. 1995; 60: 821-827. [CrossRef]
- Ullah MA, Hill GR, Tey S-K. Functional reconstitution of natural killer cells in allogeneic hematopoietic stem cell transplantation. Front Immunol. 2016; 7: 144. [CrossRef]
- Huenecke S, Cappel C, Esser R, Pfirrmann V, Salzmann-Manrique E, Betz S, et al. Development of three different NK cell subpopulations during immune reconstitution after pediatric allogeneic hematopoietic stem cell transplantation: Prognostic markers in GvHD and viral infections. Front Immunol. 2017; 8: 109. [CrossRef]
- Russo A, Oliveira G, Berglund S, Greco R, Gambacorta V, Cieri N, et al. NK cell recovery after haploidentical HSCT with posttransplant cyclophosphamide: Dynamics and clinical implications. Blood. 2018; 131: 247-262. [CrossRef]
- Soiffer RJ, Gonin R, Murray C, Robertson MJ, Cochran K, Chartier S, et al. Prediction of graftversus-host disease by phenotypic analysis of early immune reconstitution after CD6depleted allogeneic bone marrow transplantation. Blood. 1993; 82: 2216-2223. [CrossRef]
- Yamasaki S, Henzan H, Ohno Y, Yamanaka T, Iino T, Itou Y, et al. Influence of transplanted dose of CD56+ cells on development of graft-versus-host disease in patients receiving G-CSFmobilized peripheral blood progenitor cells from HLA-identical sibling donors. Bone Marrow Transplant. 2003; 32: 505-510. [CrossRef]
- Abrahamsen IW, Sømme S, Heldal D, Egeland T, Kvale D, Tjønnfjord GE. Immune reconstitution after allogeneic stem cell transplantation: the impact of stem cell source and graft-versus-host disease. Haematologica. 2005; 90: 86-93.
- Kim DH, Sohn SK, Lee NY, Baek JH, Kim JG, Won DIL, et al. Transplantation with higher dose of natural killer cells associated with better outcomes in terms of non-relapse mortality and infectious events after allogeneic peripheral blood stem cell transplantation from HLAmatched sibling donors. Eur J Haematol. 2005; 75: 299-308. [CrossRef]
- Larghero J, Rocha V, Porcher R, Filion A, Ternaux B, Lacassagne M-N, et al. Association of bone marrow natural killer cell dose with neutrophil recovery and chronic graft-versus-host disease after HLA identical sibling bone marrow transplants. Br J Haematol. 2007; 138: 101109. [CrossRef]
- Savani BN, Mielke S, Adams S, Uribe M, Rezvani K, Yong ASM, et al. Rapid natural killer cell recovery determines outcome after T-cell-depleted HLA-identical stem cell transplantation in patients with myeloid leukemias but not with acute lymphoblastic leukemia. Leukemia. 2007; 21: 2145-2152. [CrossRef]
- Tanaka M, Kobayashi S, Numata A, Tachibana T, Takasaki H, Maruta A, et al. The impact of the dose of natural killer cells in the graft on severe acute graft-versus-host disease after unrelated bone marrow transplantation. Leuk Res. 2012; 36: 699-703. [CrossRef]
- Kim SY, Lee H, Han M-S, Shim H, Eom H-S, Park B, et al. Post-transplantation natural killer cell count: A predictor of acute graft-versus-host disease and survival outcomes after allogeneic hematopoietic stem cell transplantation. Clin Lymphoma Myeloma Leuk. 2016; 16: 527-535.e2. [CrossRef]
- Jacobs R, Stoll M, Stratmann G, Leo R, Link H, Schmidt RE. CD16- CD56+ natural killer cells after bone marrow transplantation. Blood. 1992; 79: 3239-3244. [CrossRef]
- Giebel S, Dziaczkowska J, Czerw T, Wojnar J, Krawczyk-Kulis M, Nowak I, et al. Sequential recovery of NK cell receptor repertoire after allogeneic hematopoietic SCT. Bone Marrow Transplant. 2010; 45: 1022-1030. [CrossRef]
- Dulphy N, Haas P, Busson M, Belhadj S, Peffault de Latour R, Robin M, et al. An unusual CD56(bright) CD16(low) NK cell subset dominates the early posttransplant period following HLA-matched hematopoietic stem cell transplantation. J Immunol. 2008; 181: 2227-2237. [CrossRef]
- Breuer S, Preuner S, Fritsch G, Daxberger H, Koenig M, Poetschger U, et al. Early recipient chimerism testing in the T- and NK-cell lineages for risk assessment of graft rejection in pediatric patients undergoing allogeneic stem cell transplantation. Leukemia. 2011; 26: 509519. [CrossRef]
- Burek Kamenaric M, Kamenaric MB, Jankovic KS, Grubic Z, Seiwerth RS, Maskalan M, et al. The impact of KIR2DS4 gene on clinical outcome after hematopoietic stem cell transplantation. Hum Immunol. 2017; 78: 95-102. [CrossRef]
- Faridi RM, Kemp TJ, Dharmani-Khan P, Lewis V, Tripathi G, Rajalingam R, et al. Donorrecipient matching for KIR genotypes reduces chronic GVHD and missing inhibitory KIR ligands protect against relapse after myeloablative, HLA matched hematopoietic cell transplantation. PLoS One. 2016; 11: e0158242. [CrossRef]
- Littera R, Orrù N, Caocci G, Sanna M, Mulargia M, Piras E, et al. Interactions between killer immunoglobulin-like receptors and their human leucocyte antigen Class I ligands influence the outcome of unrelated haematopoietic stem cell transplantation for thalassaemia: A novel predictive algorithm. Br J Haematol. 2012; 156: 118-128. [CrossRef]
- Sahin U, Dalva K, Gungor F, Ustun C, Beksac M. Donor-recipient killer immunoglobulin like receptor (KIR) genotype matching has a protective effect on chronic graft versus host disease and relapse incidence following HLA-identical sibling hematopoietic stem cell transplantation. Ann Hematol. 2018; 97: 1027-1039. [CrossRef]
- Gaafar A, Sheereen A, Almohareb F, Eldali A, Chaudhri N, Mohamed SY, et al. Prognostic role of KIR genes and HLA-C after hematopoietic stem cell transplantation in a patient cohort with acute myeloid leukemia from a consanguineous community. Bone Marrow Transplant. 2018; 53: 1170-1179. [CrossRef]
- Ludajic K, Balavarca Y, Bickeböller H, Rosenmayr A, Fae I, Fischer GF, et al. KIR genes and KIR ligands affect occurrence of acute GVHD after unrelated, 12/12 HLA matched, hematopoietic stem cell transplantation. Bone Marrow Transplant. 2009; 44: 97-103. [CrossRef]
- Clausen J, Kircher B, Auberger J, Schumacher P, Ulmer H, Hetzenauer G, et al. The role of missing killer cell immunoglobulin-like receptor ligands in T cell replete peripheral blood stem cell transplantation from HLA-identical siblings. Biol Blood Marrow Transplant. 2010; 16: 273-280. [CrossRef]
- Sivori S, Carlomagno S, Falco M, Romeo E, Moretta L, Moretta A. Natural killer cells expressing the KIR2DS1-activating receptor efficiently kill T-cell blasts and dendritic cells: Implications in haploidentical HSCT. Blood. 2011; 117: 4284-4292. [CrossRef]
- Hu LJ, Zhao XY, Yu XX, Lv M, Han TT, Han W, et al. Quantity and quality reconstitution of NKG2A natural killer cells are associated with graft-versus-host disease after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2018. doi:10.1016/j.bbmt.2018.08.008. [CrossRef]
- Cooley S. KIR reconstitution is altered by T cells in the graft and correlates with clinical outcomes after unrelated donor transplantation. Blood. 2005; 106: 4370-4376. [CrossRef]
- Kang L, Voskinarian-Berse V, Law E, Reddin T, Bhatia M, Hariri A, et al. Characterization and ex vivo expansion of human placenta-derived natural killer cells for cancer immunotherapy. Front Immunol. 2013; 4. doi:10.3389/fimmu.2013.00101. [CrossRef]
- Choi I, Yoon SR, Park SY, Kim H, Jung SJ, Jang YJ, et al. Donor-derived natural killer cells infused after human leukocyte antigen-haploidentical hematopoietic cell transplantation: a dose-escalation study. Biol Blood Marrow Transplant. 2014; 20: 696-704. [CrossRef]
- Yoon SR, Lee YS, Yang SH, Ahn KH, Lee JH, Lee JH, et al. Generation of donor natural killer cells from CD34 progenitor cells and subsequent infusion after HLA-mismatched allogeneic hematopoietic cell transplantation: a feasibility study. Bone Marrow Transplant. 2009; 45: 1038-1046. [CrossRef]
- Curti A, Ruggeri L, D’Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood. 2011; 118: 3273-3279. [CrossRef]
- Stern M, Passweg JR, Meyer-Monard S, Esser R, Tonn T, Soerensen J, et al. Pre-emptive immunotherapy with purified natural killer cells after haploidentical SCT: A prospective phase II study in two centers. Bone Marrow Transplant. 2013; 48: 433-438. [CrossRef]
- Lee DA, Denman CJ, Rondon G, Woodworth G, Chen J, Fisher T, et al. Haploidentical natural killer cells infused before allogeneic stem cell transplantation for myeloid malignancies: A phase I trial. Biol Blood Marrow Transplant. 2016; 22: 1290-1298. [CrossRef]
- Shah NN, Baird K, Delbrook CP, Fleisher TA, Kohler ME, Rampertaap S, et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood. 2015; 125: 784-792. [CrossRef]
- Taniguchi M, Harada M, Kojo S, Nakayama T, Wakao H. The regulatory role of Valpha14 NKT cells in innate and acquired immune response. Annu Rev Immunol. 2003; 21: 483-513. [CrossRef]
- Yao Z, Liu Y, Jones J, Strober S. Differences in Bcl-2 expression by T-cell subsets alter their balance after in vivo irradiation to favor CD4+Bcl-2hi NKT cells. Eur J Immunol. 2009; 39: 763-775. [CrossRef]
- Lan F, Zeng D, Higuchi M, Huie P, Higgins JP, Strober S. Predominance of NK1.1 TCR or DX5 TCR T cells in mice conditioned with fractionated lymphoid irradiation protects against graftversus-host disease: “Natural Suppressor” cells. J Immunol. 2001; 167: 2087-2096. [CrossRef]
- Lan F, Zeng D, Higuchi M, Higgins JP, Strober S. Host conditioning with total lymphoid irradiation and antithymocyte globulin prevents graft-versus-host disease: the role of CD1reactive natural killer T cells. Biol Blood Marrow Transplant. 2003; 9: 355-363. [CrossRef]
- Liu YP, Li Z, Nador RG, Strober S. Simultaneous protection against allograft rejection and graft-versus-host disease after total lymphoid irradiation: Role of natural killer T cells. Transplantation. 2008; 85: 607-614. [CrossRef]
- Morecki S, Panigrahi S, Pizov G, Yacovlev E, Gelfand Y, Eizik O, et al. Effect of KRN7000 on induced graft-vs-host disease. Exp Hematol. 2004; 32: 630-637. [CrossRef]
- Hashimoto D, Asakura S, Miyake S, Yamamura T, Van Kaer L, Liu C, et al. Stimulation of host NKT cells by synthetic glycolipid regulates acute graft-versus-host disease by inducing Th2 polarization of donor T cells. J Immunol. 2005; 174: 551-556. [CrossRef]
- Duramad O, Laysang A, Li J, Ishii Y, Namikawa R. Pharmacologic expansion of donor-derived, naturally occurring CD4 Foxp3 regulatory T cells reduces acute graft-versus-host disease lethality without abrogating the graft-versus-leukemia effect in murine models. Biol Blood Marrow Transplant. 2011; 17: 1154-1168. [CrossRef]
- Hirai T, Ishii Y, Ikemiyagi M, Fukuda E, Omoto K, Namiki M, et al. A novel approach inducing transplant tolerance by activated invariant natural killer T cells with costimulatory blockade. Am J Transplant. 2014; 14: 554-567. [CrossRef]
- Du J, Paz K, Thangavelu G, Schneidawind D, Baker J, Flynn R, et al. Invariant natural killer T cells ameliorate murine chronic GVHD by expanding donor regulatory T cells. Blood. 2017; 129: 3121-3125. [CrossRef]
- Pierini A, Colonna L, Alvarez M, Schneidawind D, Nishikii H, Baker J, et al. Donor requirements for regulatory T cell suppression of murine graft-versus-host disease. J Immunol. 2015; 195: 347-355. [CrossRef]
- Hongo D, Tang X, Zhang X, Engleman EG, Strober S. Tolerogenic interactions between CD8+ dendritic cells and NKT cells prevent rejection of bone marrow and organ grafts. Blood. 2017; 129: 1718-1728. [CrossRef]
- Haraguchi K, Takahashi T, Matsumoto A, Asai T, Kanda Y, Kurokawa M, et al. Host-residual invariant NK T cells attenuate graft-versus-host immunity. J Immunol. 2005; 175: 1320-1328. [CrossRef]
- Leveson-Gower DB, Olson JA, Sega EI, Luong RH, Baker J, Zeiser R, et al. Low doses of natural killer T cells provide protection from acute graft-versus-host disease via an IL-4-dependent mechanism. Blood. 2011; 117: 3220-3229. [CrossRef]
- Schneidawind D, Baker J, Pierini A, Buechele C, Luong RH, Meyer EH, et al. Third-party CD4+ invariant natural killer T cells protect from murine GVHD lethality. Blood. 2015; 125: 34913500. [CrossRef]
- Kuwatani M, Ikarashi Y, Iizuka A, Kawakami C, Quinn G, Heike Y, et al. Modulation of acute graft-versus-host disease and chimerism after adoptive transfer of in vitro-expanded invariant Valpha14 natural killer T cells. Immunol Lett. 2006; 106: 82-90. [CrossRef]
- Yang J, Gao L, Liu Y, Ren Y, Xie R, Fan H, et al. Adoptive therapy by transfusing expanded donor murine natural killer T cells can suppress acute graft-versus-host disease in allogeneic bone marrow transplantation. Transfusion. 2010; 50: 407-417. [CrossRef]
- Chaidos A, Patterson S, Szydlo R, Chaudhry MS, Dazzi F, Kanfer E, et al. Graft invariant natural killer T-cell dose predicts risk of acute graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Blood. 2012; 119: 5030-5036. [CrossRef]
- Rubio M-T, Moreira-Teixeira L, Bachy E, Bouillié M, Milpied P, Coman T, et al. Early posttransplantation donor-derived invariant natural killer T-cell recovery predicts the occurrence of acute graft-versus-host disease and overall survival. Blood. 2012; 120: 21442154. [CrossRef]
- Haraguchi K, Takahashi T, Hiruma K, Kanda Y, Tanaka Y, Ogawa S, et al. Recovery of Valpha24+ NKT cells after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2004; 34: 595-602. [CrossRef]
- Kohrt HE, Turnbull BB, Heydari K, Shizuru JA, Laport GG, Miklos DB, et al. TLI and ATG conditioning with low risk of graft-versus-host disease retains antitumor reactions after allogeneic hematopoietic cell transplantation from related and unrelated donors. Blood. 2009; 114: 1099-1109. [CrossRef]
- Messina G, Giaccone L, Festuccia M, Irrera G, Scortechini I, Sorasio R, et al. Multicenter experience using total lymphoid irradiation and antithymocyte globulin as conditioning for allografting in hematological malignancies. Biol Blood Marrow Transplant. 2012; 18: 16001607. [CrossRef]
- Baron F, Zachée P, Maertens J, Kerre T, Ory A, Seidel L, et al. Non-myeloablative allogeneic hematopoietic cell transplantation following fludarabine plus 2 Gy TBI or ATG plus 8 Gy TLI: A phase II randomized study from the Belgian Hematological Society. J Hematol Oncol. 2015; 8: 4. [CrossRef]
- Andrews KJ, Neale G, Verbist K, Tedrick P, Nichols KE, Pereira S, et al. Anti-CD2/CD3/CD28 stimulation augments cytokine secretion and anti-tumor cytotoxicity of human invariant NKT cells expanded ex vivo. Biol Blood Marrow Transplant. 2018.
- Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood. 2014; 124: 2824-2833. [CrossRef]
- Schmid H, Schneidawind C, Jahnke S, Kettemann F, Secker K-A, Duerr-Stoerzer S, et al. Culture-expanded human invariant natural killer T cells suppress T-cell alloreactivity and eradicate leukemia. Front Immunol. 2018; 9: 1817. [CrossRef]
- Trujillo-Ocampo A, Cho H-W, Herrmann AC, Ruiz-Vazquez W, Thornton AB, He H, et al. Rapid ex vivo expansion of highly enriched human invariant natural killer T cells via single antigenic stimulation for cell therapy to prevent graft-versus-host disease. Cytotherapy. 2018; 20: 1089-1101. [CrossRef]
- Correia DV, Lopes AC, Silva-Santos B. Tumor cell recognition by γδ T lymphocytes. Oncoimmunology. 2013; 2: e22892. [CrossRef]
- Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB. The burgeoning family of unconventional T cells. Nat Immunol. 2015; 16: 1114-1123. [CrossRef]
- Born WK, Kemal Aydintug M, O’Brien RL. Diversity of γδ T-cell antigens. Cell Mol Immunol. 2012; 10: 13-20. [CrossRef]
- Hu Y, Cui Q, Luo C, Luo Y, Shi J, Huang H. A promising sword of tomorrow: Human γδ T cell strategies reconcile allo-HSCT complications. Blood Rev. 2016; 30: 179-188. [CrossRef]
- McCallion O, Hester J, Issa F. Deciphering the contribution of γδ T cells to outcomes in transplantation. Transplantation. 2018. doi:10.1097/TP.0000000000002335. [CrossRef]
- Bertaina A, Merli P, Rutella S, Pagliara D, Bernardo ME, Masetti R, et al. HLA-haploidentical stem cell transplantation after removal of αβ+ T and B cells in children with nonmalignant disorders. Blood. 2014; 124: 822-826. [CrossRef]
- Drobyski WR, Majewski D, Hanson G. Graft-facilitating doses of ex vivo activated gammadelta T cells do not cause lethal murine graft-vs.-host disease. Biol Blood Marrow Transplant. 1999; 5: 222-230. [CrossRef]
- Blazar BR, Taylor PA, Panoskaltsis-Mortari A, Barrett TA, Bluestone JA, Vallera DA. Lethal murine graft-versus-host disease induced by donor gamma/delta expressing T cells with specificity for host nonclassical major histocompatibility complex class Ib antigens. Blood. 1996; 87: 827-837. [CrossRef]
- Maeda Y, Reddy P, Lowler KP, Liu C, Bishop DK, Ferrara JLM. Critical role of host gammadelta T cells in experimental acute graft-versus-host disease. Blood. 2005; 106: 749-755. [CrossRef]
- Ellison CA, MacDonald GC, Rector ES, Gartner JG. Gamma delta T cells in the pathobiology of murine acute graft-versus-host disease. Evidence that gamma delta T cells mediate natural killer-like cytotoxicity in the host and that elimination of these cells from donors significantly reduces mortality. J Immunol. 1995; 155: 4189-4198.
- Drobyski WR, Vodanovic-Jankovic S, Klein J. Adoptively transferred gamma delta T cells indirectly regulate murine graft-versus-host reactivity following donor leukocyte infusion therapy in mice. J Immunol. 2000; 165: 1634-1640. [CrossRef]
- Lamb LS Jr, Henslee-Downey PJ, Parrish RS, Godder K, Thompson J, Lee C, et al. Increased frequency of TCR gamma delta + T cells in disease-free survivors following T cell-depleted, partially mismatched, related donor bone marrow transplantation for leukemia. J Hematother. 1996; 5: 503-509. [CrossRef]
- Godder KT, Henslee-Downey PJ, Mehta J, Park BS, Chiang K-Y, Abhyankar S, et al. Long term disease-free survival in acute leukemia patients recovering with increased gammadelta T cells after partially mismatched related donor bone marrow transplantation. Bone Marrow Transplant. 2007; 39: 751-757. [CrossRef]
- Cela ME, Holladay MS, Rooney CM, Richardson S, Alexander B, Krance RA, et al. Gamma delta T lymphocyte regeneration after T lymphocyte-depleted bone marrow transplantation from mismatched family members or matched unrelated donors. Bone Marrow Transplant. 1996; 17: 243-247.
- Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999; 284: 143-147. [CrossRef]
- Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006; 8: 315-317. [CrossRef]
- Bartholomew A, Sturgeon C, Siatskas M, Ferrer K, McIntosh K, Patil S, et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. 2002; 30: 42-48. [CrossRef]
- Vladimirovna IL, Sosunova E, Nikolaev A, Nenasheva T. Mesenchymal stem cells and myeloid derived suppressor cells: Common traits in immune regulation. J Immunol Res. 2016; 2016: 7121580. [CrossRef]
- Liu FD, Tam K, Pishesha N, Poon Z, Van Vliet KJ. Improving hematopoietic recovery through modeling and modulation of the mesenchymal stromal cell secretome. Stem Cell Res Ther. 2018; 9: 268. [CrossRef]
- Cho K, Kim Y, Park M, Kim HJ, Woo S, Park J, et al. Conditioned medium from human palatine tonsil mesenchymal stem cells attenuates acute graft‑vs.‑host disease in mice. Mol Med Rep. 2018. doi:10.3892/mmr.2018.9659. [CrossRef]
- Castello LM, Leone M, Adamini A, Castiglia S, Mareschi K, Ferrero I, et al. Analysis of mesenchymal stromal cell engraftment after allogeneic HSCT in pediatric patients: A large multicenter study. J Pediatr Hematol Oncol. 2018; 40: e486-e489. [CrossRef]
- Berger M, Mareschi K, Castiglia S, Rustichelli D, Mandese A, Migliore E, et al. In vitro mesenchymal progenitor cell expansion is a predictor of transplant-related mortality and acute GvHD III-IV after bone marrow transplantation in univariate analysis: A large singlecenter experience. J Pediatr Hematol Oncol. 2019; 41: 42-46. [CrossRef]
- Wang ZK, Yu HJ, Cao FL, Liu ZH, Liu ZY, Feng WJ, et al. Donor-derived marrow mesenchymal stromal cell co-transplantation following a haploidentical hematopoietic stem cell transplantation trail to treat severe aplastic anemia in children. Ann Hematol. 2018. doi:10.1007/s00277-018-3523-2. [CrossRef]
- Ball LM, Bernardo ME, Roelofs H, Lankester A, Cometa A, Egeler RM, et al. Cotransplantation of ex vivo expanded mesenchymal stem cells accelerates lymphocyte recovery and may reduce the risk of graft failure in haploidentical hematopoietic stem-cell transplantation. Blood. 2007; 110: 2764-2767. [CrossRef]
- Wang L, Zhu CY, Ma DX, Gu ZY, Xu CC, Wang FY, et al. Efficacy and safety of mesenchymal stromal cells for the prophylaxis of chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation: a meta-analysis of randomized controlled trials. Ann Hematol. 2018; 97: 1941-1950. [CrossRef]
