

Telomere Editorial - Perspectives on Telomeres and Aging
Michael Fossel * ![]()
- Telocyte LLC, 250 Monroe NE, Grand Rapids, MI, USA
* Correspondence: Michael Fossel ![]()
Special Issue: Perspectives on Telomeres and Aging
Received: January 29, 2019 | Accepted: January 30, 2019 | Published: January 31, 2019
OBM Geriatrics 2019, Volume 3, Issue 1 doi:10.21926/obm.geriatr.1901031
Recommended citation: Fossel M. Telomere Editorial - Perspectives on Telomeres and Aging. OBM Geriatrics 2019; 3(1): 031; doi:10.21926/obm.geriatr.1901031.
© 2019 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
The lack of research on the cause of aging is no less than a scandal because it is ignored by the medical establishment who ironically argue that aging is the greatest risk factor for their favorite disease.
-- Leonard Hayflick, 2019, Personal Communication
Beginning in the 1960’s [1,2], cell senescence was first suggested as a central facet in aging, and therefore in age-related disease. In the early 1990’s [3,4,5], key papers suggested that cell senescence was controlled by telomere shortening and that this correlated with age-related disease [6]. This was shown to be not merely a correlational, but a causal relationship in the landmark paper of 1998 [7], which showed that resetting telomere lengths could reset cell aging. This was followed thereafter by a series of papers [8,9,10] showing that this could be extended to in vitro demonstration of regrowth of young tissue from old cells or prolonged maintenance of young cell characteristics in skin, bone, and vascular tissue.
The first publications outlining the clinical potential of this field appeared in late 1990-2000 [11,12,13,14], followed by the only textbook on this area [15], which described not only the genetic and cellular processes involved in age-related disease, but reviewed age-related human pathologies tissue-by-tissue, explaining both the probable role of cell senescence and the potential for more effective clinical intervention. The overall theory has been reviewed since [16] and a more recent book [17] brought the field up-to-date, outlining the current status of the field and the practical prospects for human clinical trials.
In addition, a number of papers [18,19] have demonstrated the success of using telomerase therapy in animals, resulting in extended healthy lifespans and, perhaps more importantly, strong support for the belief that we may more effectively intervene in age-related human diseases. As a result of these papers and ones like them, FDA-sanctioned telomerase gene therapy trials are planned.
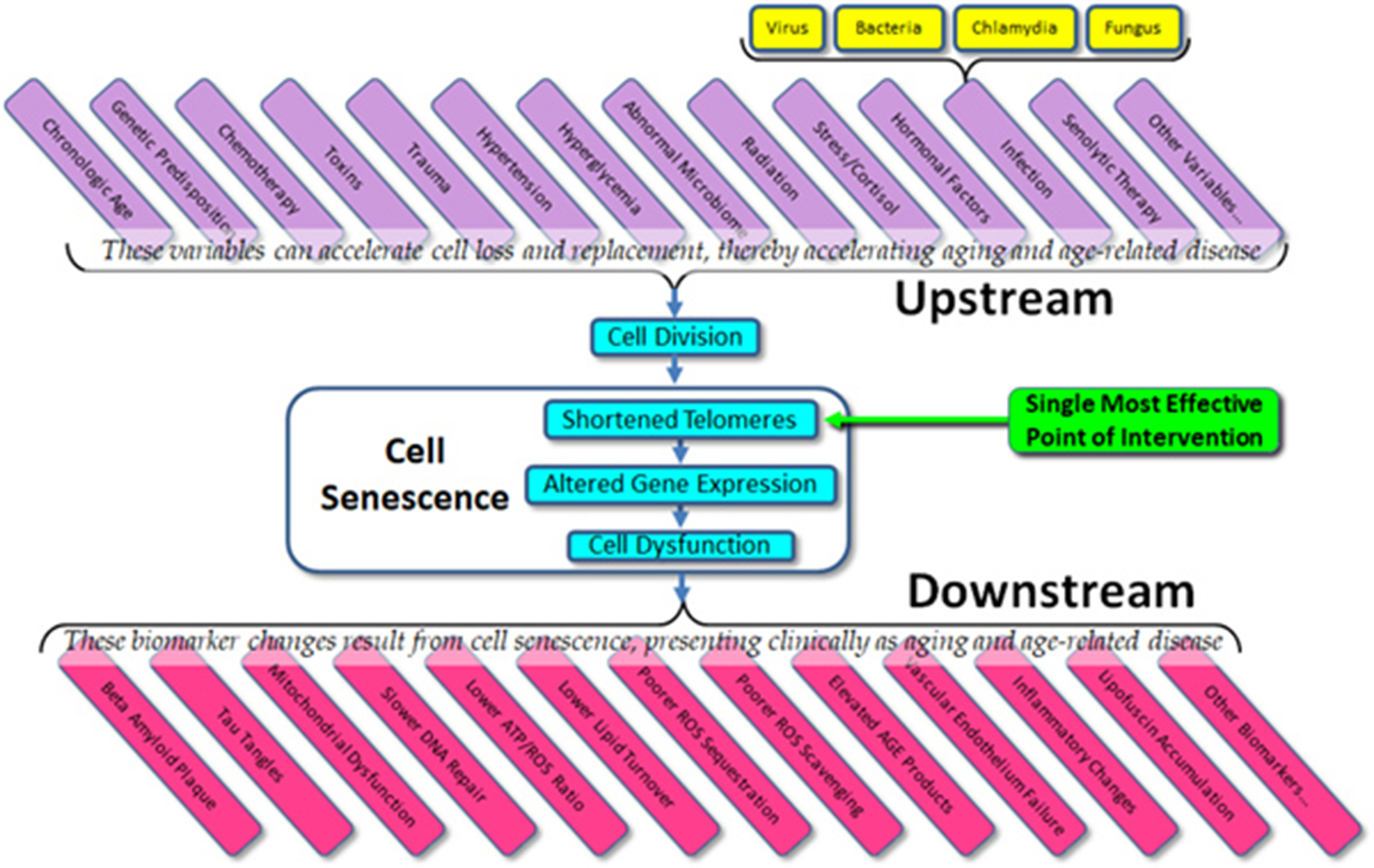
Despite this history and its clinical potential, the model is not widely understood, and misconceptions are common, even within the academic literature. The model suggests a complex cascade of causation, which might be simplified as seen in Figure 1. We might summarize this model as: 1) cells divide, 2) telomeres shorten, 3) epigenetic changes result, and 4) cells show subtle but pervasive detrimental effects in their function. Such dysfunctional changes in cell function are ubiquitous. As examples, the turnover rate of most protein pools (e.g., enzymes, structural proteins, etc.) slows down, resulting in a gradual increase in the percentage of denatured (i.e., ineffective) proteins. As one example, the four major families of DNA repair enzymes become less effective, resulting in slower and less effective DNA repair, with a consequent increase in the percentage of mutational changes at any one time. In the mitochondria, the major aerobic enzymes are likewise turned over more slowly, decreasing the efficiency of the mitochondria and increasing the ROS/ATP ratio. Moreover, lipid membrane molecules – as they become oxidized or otherwise denatured – are more slowly replaced, culminating in leakier and less effective membrane barriers, with the result that (for example) ROS, the majority of which are created in the mitochondria, are more likely to traverse the mitochondrial walls and result in damage to molecules within the cytosol and nucleus. This, coupled with slower turnover among scavenger proteins (e.g., SOD, catalase, etc.) and a lower rate of turnover of damaged molecules in general, results in a gradual increase in oxidative damage to the cell.
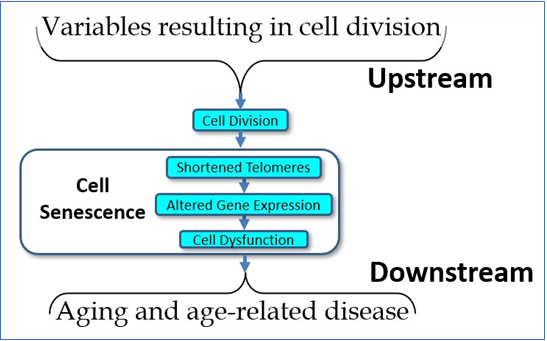
Figure 1 The simple model of telomere aging and disease.
The upshot is that it is telomere shortening and – more importantly – the gradual changes in gene expression induced by such shortening – that results in cell aging and declining cell function results in a secondary loss of tissue function. Not only are intracellular molecules less likely to undergo turnover, but the same is true of extracellular molecules, such as collagen, elastin, amyloid, etc. The result of such slower extracellular molecular turnover not only results in such relatively harmless age-related changes as wrinkles (due to the slower turnover of collagen and elastin), but the tragic and disastrous changes of Alzheimer’s disease (partially due to the slower turnover of amyloid proteins). In all cases, molecular pools within the body – whether intracellular or extracellular – are dynamic. Molecules are constantly being turned over and replaced. While different molecular pools undergo turnover at different rates, all molecular pools slow their rate of turnover with age, and the result is what we see as aging and age-related disease.
This model has not only proven consistent with all known data on aging and age-related disease, it has also proven predictively valid in trials of various interventions, such as those aimed at beta amyloid metabolism (e.g., Solanezumab, Aducanumab, etc.) mitochondrial function, or other interventional trials in the case of Alzheimer’s disease. Finally, this model has not only withstood the tests of time but is eminently testable. When tested in animals (as cited above), interventions aimed at resetting gene expression – specifically, using telomerase expression – have been consistently shown to be effective in addressing age-related changes.
Several cautions remain. The first is that one common misunderstanding of the model is to assume that cell aging is a matter of telomere length, rather than relative telomere length. The critical variable is not telomere length but change in length. While some mice have telomeres that are 10x those of most humans, their lifespans are approximately one fortieth of most humans. This is entirely consistent with the model, which predicts that cell aging is a product of relative telomere loss, not absolute telomere length. More importantly, the key feature is the alteration that occurs in gene expression – epigenetic change – which is driven by telomere loss.
A second caution is the all-too-common use of LTL’s (lymphocyte telomere lengths) as a valid measure of organismal aging, which is questionable, if not unwarranted [20,21]. While lymphocytic telomere loss is correlated with age, there are several problems with using LTL’s as a dependent variable. The first problem should be obvious: lymphocytes are only one cell type among many and are scarcely the best indicator of what is happening in other cells, tissues, and organs. If, for example, we wish to get an indication of the risk of age-related vascular disease, the valid measure would be the telomere lengths of vascular endothelial cells, not the telomere lengths of lymphocytes. LTL’s reflect the telomere lengths of such endothelial cells only roughly, in the sense that all somatic cells that divide are showing telomere loss correlating with age, and hence with one another. While true, humans die from specific age-related diseases and the status of one organ is not always an indication of the status of another. For example, your risk of heart attack may correlate with your risk of osteoarthritis (in that both correlate with chronologic age), but the best biomarker of heart attack risk is not the state of your chondrocytes, but the state of your arterial endothelial cells. A second problem with LTL’s is that circulating lymphocytes represent only a small percent of the body’s lymphocyte population, most of which are not in circulation. Worse yet, and depending on lymphocyte type (e.g., B cell, T cell, etc.) the major part of the loss of telomere length may vary in location from marrow to circulation depending upon stress to the organism. In practice, this means that there may be only minimal difference between the telomere lengths of the hematopoetic stem cells and circulating lymphocytes when there is little or no stress to the immune system, but a great deal of difference between stem cells and circulating lymphocytes when the immune system is stressed. The practical upshot is that LTL’s do not provide an accurate indication of the “age” of the hematopoetic system except transiently when peripheral turnover is the exception.
Putting it simplistically, the telomere lengths of hematopoetic stem cells (which are rarely measured) show a very gradual and predictable decline with age, while peripheral lymphocytes are more likely to show a variable and unpredictable change over time, depending upon the needs of and the environment of the organism (see Figure 2). As LTL’s may (and do) vary due to multiple factors, they are neither valid nor reliable indicators of the physiological age of the organism. They may be useful, but only when interpreted with great caution and then only in context. A final problem is that cell function (and the increasing dysfunction of senescing cells) correlates well with the minimal, rather than the mean telomere length, yet most studies (even when they identify their methods clearly) rely upon the cheaper, but less relevant mean telomere length, thus invalidating their conclusions.
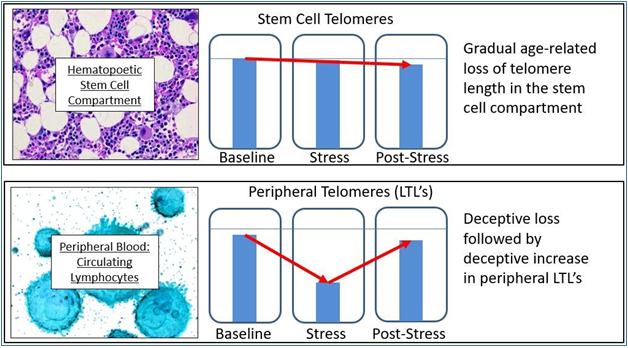
Figure 2 The problem with using LTL’s.
The upshot of these several issues is that most current studies that purport to evaluate telomere lengths and assess potential variables or interventions, measure an invalid and unreliable variable and do so in an invalid and unreliable manner. While the conclusions of such studies may (perchance) be correct, the data used in these studies is insufficient support for the stated conclusions. As examples, the use of dietary improvements, exercise programs, meditation, or medication have all been suggested as potentially able to elongate telomeres (and improve health via this mechanism), but there is no adequate data supporting any of these conclusions.
The current state of the field – the importance to telomeres and telomerase in aging and age-related disease – is confused and open to question, but the potential remains as enormous as it was suggested to be 20 years ago. Given what we do know about cell senescence, telomeres, and age-related disease, we might list both the obvious variables that affect cell aging (“upstream”) and the dependent variables that result from cell aging (“downstream”) (Figure 3). What is immediately apparent is that in almost all cases, current therapeutic approaches (and experimental trials) are targeted at downstream biomarkers, rather than targeting upstream variables or telomeres themselves. As all of the upstream variables affect telomere length (and gene expression) and as we are now capable of elongating telomeres in vivo, the single most effective point of intervention is not the dozens of upstream variables and certainly not the downstream biomarkers, but telomere length per se. Attempts to treat age-related diseases by addressing downstream biomarkers – such as targeting amyloid, tau proteins, mitochondria, or inflammation in the case of Alzheimer’s disease – have not only proven uniformly ineffective, but we can now see why these approaches have been (and will continue to be) ineffective. Such approaches are essentially treating symptoms rather than treating the disease itself.
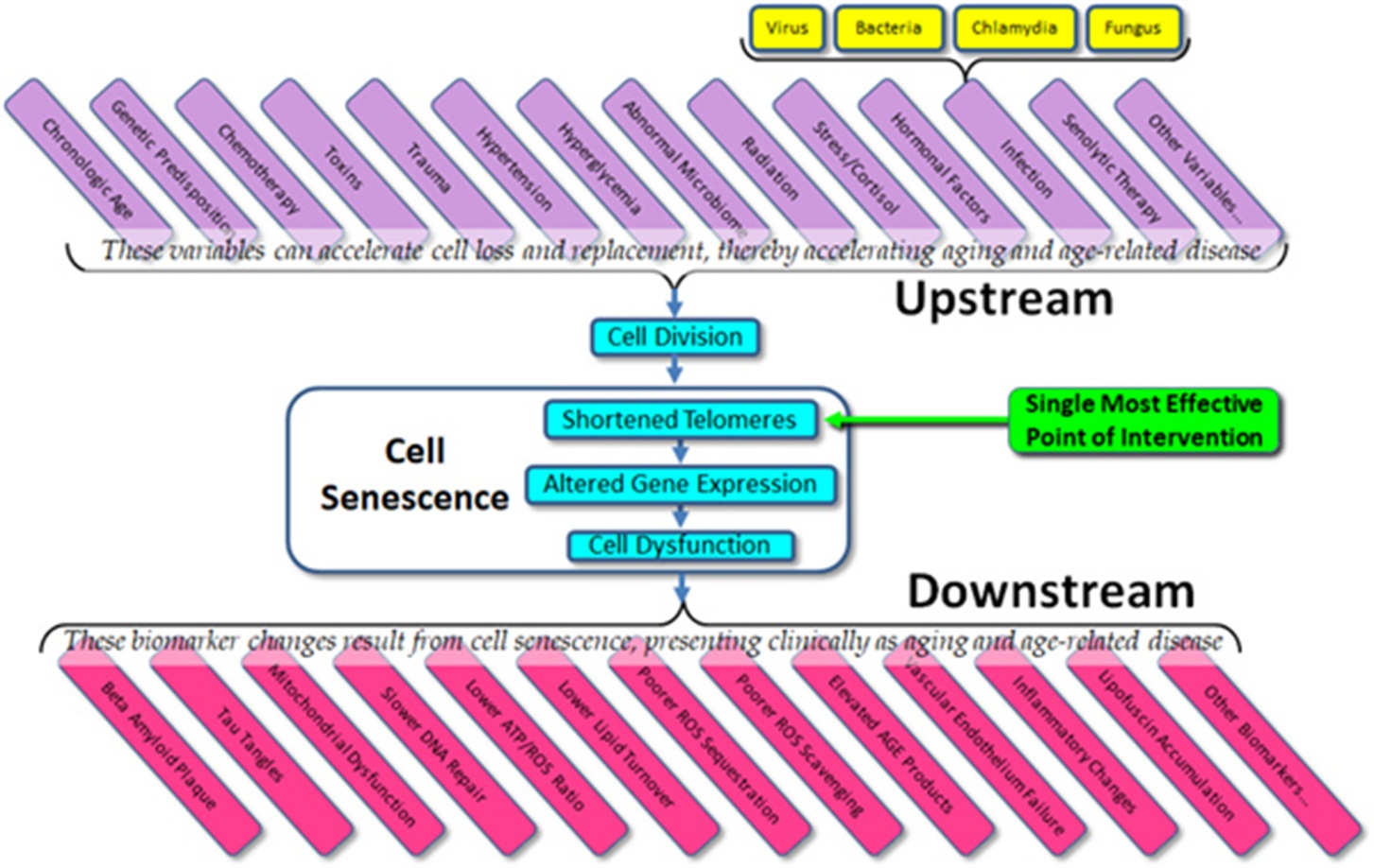
Figure 3 Upstream variables and downstream biomarkers in aging and disease.
Approaches that do not target telomere length, such as ablation of senescing cells will likewise prove futile, if not actually damaging (see related article in this issue). In cases in which we have targeted telomere length directly, not only do we find reliable improvement in all measured downstream biomarkers as well as general organismal health (and lifespan), but despite early concerns in the literature, there has been no evidence of cancer or other adverse effects [19,22].
Understanding the role of cell senescence, telomeres, and telomerase in aging and age-related disease remains the most promising avenue for therapeutic intervention. The clinical potential of telomerase gene therapy is without any equal in current therapeutic approaches.
Author Contributions
The author wrote this paper.
Competing Interests
The author has declared that no competing interests exist.
References
- Hayflick L, Moorhead PS. The serial cultivation of human diploid strains. Exp Cell Res. 1961; 25: 585-621. [CrossRef]
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965; 37: 614-636. [CrossRef]
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during aging of human fibroblasts. Nature. 1990; 345: 458-460. [CrossRef]
- Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA. 1992; 89: 10114-10118. [CrossRef]
- Allsopp RC, Chang E, Kashefi Aazam M, Rogaev EI, Piatyszek MA, Shay JW, et al. Telomere shortening is associated with cell division in vitro and in vivo. Exp Cell Res. 1995; 220: 194 200. [CrossRef]
- Chang E, Harley CB. Telomere length and replicative aging in human vascular tissues. Proc Nat Acad Sci USA. 1995; 92: 1190-11194. [CrossRef]
- Bodnar AG, Chiu C, Frolkis M, Harley CB, Holt SE, Lichtsteiner S, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998; 279: 349-352. [CrossRef]
- Funk WD, Wang CK, Shelton DN, Harley CB, Pagon GD, Hoeffler WK. Telomerase expression restores dermal integrity to in vitro-aged fibroblasts in a reconstituted skin model. Exp Cell Res. 2000; 258: 270-278. [CrossRef]
- Yudoh K, Matsuno H, Nakazawa F, Katayama R, Kimura T. Reconstituting telomerase activity using the telomerase catalytic subunit prevents the telomere shortening and replicative senescence in human osteoblasts. J Bone Miner Res. 2001; 16: 1453‑1464. [CrossRef]
- Matsushita H, Chang E, Glassford AJ, Cooke JP, Chiu CP, Tsao P. eNOS activity is reduces in senescent human endothelial cells. Preservation by hTERT immortalization. Circ Res. 2001; 89: 793‑798. [CrossRef]
- Fossel M. Reversing Human Aging. New York, William Morrow & Co. 1996.
- Banks DA, Fossel M. Telomeres, cancer, and aging; Altering the human lifespan. JAMA. 1997; 278: 1345-1348. [CrossRef]
- Fossel M. Telomerase and the aging cell; Implications for human health. JAMA. 1998; 279: 1732-1735. [CrossRef]
- Fossel M. Cell senescence and human aging: A review of the theory. In Vivo. 2000; 14: 29-34.
- Fossel M. Cells, Aging, and Human Disease. New York: Oxford University Press; 2004.
- Fossel M. Cell senescence in human aging and disease. Annals NY Acad Sci. 2006; 959: 14-23. [CrossRef]
- Fossel M. The Telomerase Revolution. Dallas: BenBella Books; 2015.
- Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams A, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase deficient mice. Nature. 2011; 469: 102-106. [CrossRef]
- de Jesus BB, Vera E, Schneeberger K, Tejera AM, Ayuso E, Bosch F, et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med. 2012; 4: 691-704. [CrossRef]
- Fossel M. Use of telomere length as a biomarker for aging and age-related disease. Curr Tran Geriatr Gerontol Rep. 2012; 1: 121–127. [CrossRef]
- Mensa E, Latini S, Ramini D, Storci G, Bonafe M, Olivieri F. The telomere world and aging: Analytical challenges and future perspectives. Ageing Res Rev. 2019; 50: 27-42. [CrossRef]
- Muñoz-Lorente MA, Martinez P, Tejera A, Whittemore K, Moises-Silva AC, Bosch F, et al. AAV9-mediated telomerase activation does not accelerate tumorigenesis in the context of oncogenic K-Ras-induced lung cancer. PLoS Genet. 2018; 14: e1007562. [CrossRef]
