

Therapies for Childhood Polycystic Kidney Disease
Ameya Patil 1 ![]() , William E Sweeney, Jr 1
, William E Sweeney, Jr 1 ![]() , Ellis D. Avner 1, 2, *
, Ellis D. Avner 1, 2, * ![]()
- Medical College of Wisconsin, Department of Pediatrics, Children’s Research Institute, Children’s Hospital Health System of Wisconsin, Milwaukee, Wisconsin 53226, USA
- Medical College of Wisconsin, Department of Physiology, Children’s Research Institute, Children’s Hospital Health System of Wisconsin, Milwaukee, Wisconsin 53226, USA
* Correspondence: Ellis D. Avner ![]()
Received: October 7, 2018 | Accepted: December 18, 2018 | Published: December 24, 2018
OBM Genetics 2018, Volume 2, Issue 4 doi: 10.21926/obm.genet.1804056
Academic Editor: Khue Vu Nguyen
Special Issue: Treatment of Genetic Disease
Recommended citation: Patil A, Sweeney WE, Avner ED. Therapies for Childhood Polycystic Kidney Disease. OBM Genetics 2018;2(4):056; doi:10.21926/obm.genet.1804056.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Renal cysts are present in a wide variety of hereditary renal diseases in children. The term polycystic kidney disease (PKD) refers to two specific hereditary diseases, distinguished by the usual age of onset and genetic cause: autosomal recessive polycystic kidney disease/congenital hepatic fibrosis (ARPKD/CHF, MIM *606702) and autosomal dominant polycystic disease (ADPKD-OMIM *601313 and OMIM *173910). ARPKD/CHF is characterized by cystic dilations of the renal collecting ducts and developmental defects of biliary ductal plate remodeling, resulting in varying degrees of congenital hepatic fibrosis. ARPKD/CHF is commonly diagnosed in utero or at birth but can remain silent well into adolescence and rarely into adulthood. ADPKD, the most common inherited renal disease is characterized by slow, progressive enlargement of fluid-filled cysts leading to renal failure by the fifth to sixth decade of life in addition to various extrarenal manifestations. ADPKD can manifest in utero, infants, and children and can be a significant cause of morbidity and mortality in this age group. Our understanding of the genetic basis of ARPKD and ADPKD, including mechanisms of transmission and genes involved continues to evolve. Despite remarkable advances in understanding the basic molecular and cellular pathophysiology, the development of targeted therapies has proven difficult.
The complex structure of the genes and proteins, the tremendous allelic heterogeneity, complex inheritance patterns, and functional interactions of both the genes and proteins products of ARPKD and ADPKD complicate diagnostic and prognostic assessments. None the less, multiple therapeutic interventions are being tested in clinical trials, two clinical trials aimed at pediatric PKD patients are underway and one therapy, tolvaptan, has been approved for adult use in Japan, Europe, Australia, South Korea and recently in the USA. This review will discuss: 1) the clinical manifestations and genetics of childhood PKD; 2) how our understanding of the molecular and cellular pathophysiology of PKD is guiding current therapies; and 3) how recent discoveries may shape future therapies.
Keywords
ARPKD; ADPKD; proliferation; therapy; tyrosine kinase inhibitor; tolvaptan; combination therapy; multi-kinase inhibitors; gene dosage
1. ARPKD/CHF
1.1 Clinical Manifestations
ARPKD/CHF is a congenital dual organ fibrocystic disease and is a significant cause of renal and liver-related morbidity and mortality in children. ARPKD/CHF previously referred to as infantile PKD, is a rare disease commonly diagnosed in utero or at birth, and occurs at a frequency of 1:20,000 live births [1]. ARPKD/CHF is characterized by non-obstructive fusiform dilations of the renal collecting ducts and varying degrees of biliary ectasia due to developmental biliary ductal plate malformations leading to congenital hepatic fibrosis [2,3,4,5]. ARPKD is highly variable with a wide clinical spectrum of disease that ranges from the severe classical presentation in utero, or at birth, of massively enlarged bilaterally cystic kidneys to the presentation in older children, teenagers, or young adults with manifestations of hepatic disease and negligible renal disease [4,6]. The variability of organ involvement in ARPKD is not well understood, but it is widely acknowledged [7,8,9,10].
The renal disease in ARPKD is characterized by nephromegaly, hypertension, and varying degrees of renal insufficiency. The most severely affected children exhibit a characteristic “Potter’s” phenotype with enormously enlarged echogenic kidneys, up to 10x the normal size, and pulmonary hypoplasia due to oligohydramnios and a rigid contraction of the diaphragm [10]. Pulmonary hypoplasia, as the result of oligohydramnios, is a serious complication that can lead to respiratory failure that may ultimately be incompatible with survival [10,11,12]. The perinatal mortality due to pulmonary hypoplasia is estimated to be as high as 30% despite advances in neonatal care [2,13].Patients that survive the neonatal period have a one-year survival rate of 85% and a ten-year survival rate of 82% [10,12,14]. The majority of patients (70-80%) manifest some degree of impaired renal function just after birth, but function stabilizes as renal development continues and the kidney may get smaller [10,15,16]. Most patients have a urinary concentrating defect with consequent polyuria and polydipsia which can lead to clinically significant electrolyte abnormalities [10,15,17,18,19].
Neonatal survivors and patients diagnosed as older children or adults typically present with signs and symptoms related to CHF [4]. Histological evidence of CHF is invariably present at birth, but the clinical complications of CHF can develop at any time [3,4,20]. Portal hypertension (PH), the predominant manifestation of CHF is potentially lethal, and nearly 70% of children with ARPKD will develop PH [4,6]. PH may lead to hypersplenism with low platelet counts, and varices within the esophagus, stomach, and rectum. Rupture of esophageal or gastric varies can lead to a life-threatening hemorrhage [4,10].
A subset of affected individuals will develop recurrent or persistent bacterial ascending cholangitis due to dilated bile ducts and stagnant bile flow [4,10,21]. Patients may require portosystemic shunting or liver transplantation for complications of portal hypertension [10,22].
The variability in age and clinical manifestations of the hepatic disorder is related to the relative degree of renal and biliary abnormalities [2,23]. In neonatal survivors, estimates are that 40% will exhibit severe dual-organ disease, and the remaining 60% is divided equally into three groups: those with severe renal disease/mild hepatobiliary disease; mild renal/severe hepatobiliary kidney disease; mild renal/mild hepatobiliary disease: [4,22]. Platelet counts are an accurate means of determining the severity of PH in patients with ARPKD [4,20].
Hypertension is common in both infants and children, and it can be challenging to control [3,24,25,26]. Nearly all children with ARPKD will develop hypertension including patients with normal renal function [9,25,27]. Combination ACEi/ angiotensin receptor blocker therapy is not recommended because of an increased risk of side effects without a clear-cut benefit in reducing hypertension [28]. The use of ACEi in older adolescents that may become pregnant is contraindicated due to concerns of teratogenicity [29].
Kidney transplantation offers definitive renal replacement therapy in children with ARPKD. Successful kidney transplantation prolongs survival and often accelerates growth and development in young uremic children. Patients suffering from both severe renal and severe hepatic disease may be candidates for combined liver and kidney transplant [22]. A decision tree that guides appropriate choices has been developed and published [22].
Given the multiorgan effects of ARPKD/CHF, a team of pediatric subspecialists including nephrologist, hepatologist, specialized nurses, dietitians, social workers is recommended for optimal comprehensive care for children with ARPKD [9,10].
1.2 Genetics-ARPKD
The majority of ARPKD/CHF occurs due to mutations in a single gene, the polycystic kidney and hepatic disease 1 (PKHD1) gene [30]. As the name of the gene and the disease imply, ARPKD/CHF patients invariably have some degree of CHF along with cystic renal disease [2]. The variability of renal and hepatic disease previously noted does not appear to be related to the type or site of mutations in PKHD1 [5,31].
ARPKD/CHF was originally thought to be a genetically homogeneous disease caused by mutations in a single gene until the recent discovery of a second locus, the DZIP1L gene (Daz interacting protein 1-like (OMIM *617570). Mutations in DZIP1L have been identified in seven children from four unrelated consanguineous families [32]. All patients had enlarged hyperechogenic kidneys, arterial hypertension, and four progressed to end-stage renal disease (ESRD). In animal models, mutations in DZIP1L appears to compromise membrane translocation of the ADPKD proteins, polycystin-1 and polycystin-2 (see ADPKD Proteins below). The number of ARPKD/CHF cases caused by mutations in DZIP1L is a minute percentage of a rare disease, and nearly all cases are due to PKHD1.
PKHD1, the causative gene of ARPKD/CHF, is an exceptionally large gene of 86 exons spanning 470 kb of genomic DNA with 67 exons included in the longest open-reading frame [33]. Alternatively spliced transcripts including some that may be secreted have been identified; however, the function and effect of these isoforms are not known [33,34]. Attempts to define genotype-phenotype correlations and predict disease outcomes have largely been unsuccessful. To date, only general correlations are possible at best due to extreme allelic heterogeneity [35,36]. Nearly 750 mutations in PKHD1 have been identified to date (http://www.humgen.rwth-aachen.de) without apparent grouping at any specific site [37,38,39]. The majority of families have unique (“private”) mutations, and the majority of families are compound heterozygotes [8,37,40]. Missense, frameshift and truncating mutations have all been described [41].
Patients with two truncating mutations generally exhibit a very severe phenotype with a high rate of perinatal /neonatal mortality [8,37,42]. In contrast, patients harboring at least one missense mutation (amino acid substitutions), have a non-lethal presentation [43,44]. In an animal model of Pkhd1, investigators found 22 novel renal transcripts and observed an assortment of alternative splicing mechanisms including exon skipping, use of alternate acceptor/donor splice sites, and the inclusion of novel exons [34]. These studies suggest that aberrant PKHD1 splicing may represent an additional pathogenic mechanism in ARPKD.
Finally, several phenocopies exist that can lead to a misdiagnosis of ARPKD. A recent study of 36 unrelated patients originally diagnosed with ARPKD but without an identified mutation in PKHD1 found that 22% harbored mutations in other genes including PKD1, HNF1b, NPHP1, TMEM67, and PKD1/TSC2) [45].
1.3 ARPKD Proteins
PKHD1 encodes a very large, complex protein of unknown function known as fibrocystin or polyductin (FPC) [33,40]. The longest open reading frame is comprised of 66 exons resulting in a protein of 4074 amino acids [46]. FPC is predicted to have a single transmembrane spanning domain and multiple copies of an immunoglobulin-like plexin-transcription-factor domain.Alternatively, spliced transcripts including some that may be secreted have been identified; however, the function and significance of these isoforms are unknown [33,34]. During normal renal development, FPC is strongly expressed in branching ureteric bud. Postnatally it is normally expressed in collecting ducts with reduced amounts expressed in proximal and distal nephron segments [47]. FPC is expressed in multimeric complexes on the apical and basolateral plasma membrane and in the primary cilium/basal bodies. FPC has been found in the same protein complex as the PKD2 protein PC2 [47,48].
FPC undergoes regulated proteolytic processing similar to NOTCH and releases an intracellular C-terminal fragment which translocates to the nucleus [49].
There is also evidence that FPC undergoes nonsense-mediated alternative splicing. Nonsense-mediated mRNA decay (NMD) is a mRNA quality control mechanism meant to lessen the harmful effects of truncating mutations. A shortened, truncated protein with some residual function is produced, which may yield a milder phenotype than generally expected [46,50]. FPC is also normally expressed in the liver, pancreas, and lungs and its precise function in these organs is unknown as well.
Overall, the large size of PKHD1 and its protein FPC, its complex pattern of splicing, allelic heterogeneity and lack of an accurate paradigm to identify changes as pathogenic, pose substantial challenges to DNA-based diagnostic testing. Despite these challenges, there is a robust demand for early, reliable prenatal diagnosis, for couples considering pre-implantation genetic diagnosis and possible in vitro fertilization. PDG-IVF has been successfully utilized in at-risk families [51]. Given the levels of complexities, genetic counselors should be necessary participants in any discussions and planning based on genetic sequence.
2. ADPKD
2.1 Clinical Manifestations
ADPKD is the most common potentially lethal monogenetic renal disease occurring at a frequency between 1/400 and 1/1000 [52,53]. ADPKD affects over 600,000 individuals in the USA and greater than 12.4 million people worldwide [54]. ADPKD is generally a late-onset (adult), systemic disease characterized by persistent development and unrelenting growth of bilateral focal renal cysts that eventually compresses normal renal parenchyma. This results in fibrosis and end-stage renal disease (ESRD). Nearly 50% of affected individuals will reach ESRD by the sixth decade of life [52,55].
ADPKD has extraordinarily high phenotypic variability. Despite the classic presentation as an adult disease, ADPKD can have clinical manifestations including structural kidney disease, in infants, children and young adults [3,10,56,57,58]. As many as 5% of all ADPKD patients present in childhood with a broad phenotypic spectrum, ranging from severe neonatal presentations mimicking ARPKD to renal cysts that remain clinically silent well into adulthood [58,59,60,61]. Additionally, as many as 17% of children and adolescents with ADPKD will exhibit unilateral cystic kidneys at initial presentation [10].
ADPKD is a systemic disease with extrarenal manifestations that can include: hepatic cysts; splenic cysts; pancreatic cysts; cardiac abnormalities and vascular abnormalities including intracranial aneurysms. Many of these adult extrarenal manifestations are uncommon in children with ADPKD. The incidence of hepatic cysts increases with age. Early studies of ultrasound screening determined that 10 to 20 percent of ADPKD patients below the age of 30 had detectable hepatic cysts [62,63]. However, in a study of 230 ADPKD patients between 15 and 46 years old, the use of MRI identified hepatic cysts in 83 percent of patients with ADPKD [64].
Pancreatic and splenic cysts can occur in childhood ADPKD, but the occurrence is rare and generally of no clinical consequence. There has been a single report of a ruptured intracranial aneurysms (ICA) in a patient under 20 years of age, but routine screening is not indicated during childhood [65]. A growing body of evidence suggests that hypertension, cardiovascular abnormalities, and increased kidney volume precede the development of symptoms in affected children [66,67]. Furthermore, studies have shown that targeting modifiable risk factors early in the disease can significantly slows progression [58,66,67,68]. Consequently, clinicians should be aware of these factors and pediatric patients with ADPKD should be monitored closely.
Renal manifestations in children with ADPKD are comparable to renal manifestations in affected adults [58]. In adult ADPKD, creatinine levels rise only in the advance stages of disease after non-cystic parenchyma has been permanently destroyed by expanding renal cysts. Thus, the renal function remains normal well into disease progression in adults. Similarly, renal function in children with ADPKD is generally normal [69].
Renal manifestations in children and adults with ADPKD include hypertension, hypertension induced cardiac disease, hematuria, proteinuria, polyuria, renal stones, urinary tract infections, and acute or chronic flank and abdominal pain [55,70]. Hematuria can occur with cyst hemorrhage, and overt proteinuria is generally associated with more advanced structural kidney disease [69]. There is little to no correlation between microalbuminuria and either kidney volume or function in children with ADPKD. However, studies link established proteinuria in children with a more aggressive course of the renal disease [69,71]. Glomerular hyperfiltration is common and indicates an increased risk of progression [72] including a faster decline in eGFR and a higher increased renal enlargement [73].
Hypertension induced cardiovascular disease is a major feature in both adults and children with ADPKD. Up to 30% of children with ADPKD have overt hypertension. Hypertension is particularly common in children with very early onset ADPKD [66,74,75]. Hypertension is linked to increased total kidney volume and an accelerated rate of renal function decline and left ventricular hypertrophy (LVH) [76,77]. Studies imply that a substantial degree of left ventricular hypertrophy (LVH) may be present in children with ADPKD, even those with normal BP [66,78].
Tight regulation of BP in normotensive children to the 50th percentile was found to halt the progression of LVH and slow the decline in renal function [66,79,80,81]. A similar benefit of rigorous BP control (<110/75 mmHg) on kidney volume was confirmed in the HALT-PKD study in adults. ACE inhibition monotherapy in early ADPKD reportedly results in a 14% reduction in total kidney volume growth [68].
A five-year prospective clinical trial designed to evaluate the effectiveness of angiotensin-converting enzyme inhibitor (ACEI) enalapril on BP control provided additional evidence of the critical importance of BP in children with ADPKD. This study found that ACEi treatment in normotensive (BP 75 to 95th percentile) or borderline hypertensive (BP ≤75 percentile) children with ADPKD could prevent or significantly reduce the development of LVH and the decline in renal function [71]. These benefits occurred in the absence of any positive effect on kidney volume [58,68,71]. Surprisingly, these secondary benefits of ACEi did not apply to hypertensive ADPKD children [71]. The strong correlation between hypertension, larger kidneys and cardiovascular disease in ADPKD children makes accurate and frequent monitoring of BP essential. [77,80,82,83].
Ambulatory BP monitoring (ABPM) is the most accurate method of detecting hypertension in children [84,85]. Studies of pediatric ADPKD patients assessed by ABPM have revealed: a) a significant relationship between renal volume, renal length, and number of renal cysts [86]; b) one-third of the hypertensive ADPKD children showed exclusively nocturnal hypertension [86,87]; and c) nearly 60% of patients who had normal BP by casual measurement were found to have daytime hypertension by ABPM [86]. Similar to the previously noted studies in childhood ARPKD [88], routine ABPM is more sensitive for the diagnosis of hypertension in the pediatric population with ADPKD [75,86].
Structural kidney disease can be seen in children with ADPKD and in rare instances can be detected in utero [3,56,57,58]. Structural kidney disease can cause acute or chronic flank pain and may be severe enough to necessitate chronic pain management [58].
In general, the renal manifestations seen in adult ADPKD are also seen in children except for renal failure which rarely occurs in children. It is clear that structural renal damage occurs before most children are diagnosed [66,77,79,89]. The benefits from early and rigorous BP control should prompt clinicians to monitor at-risk children regularly with ABPM to identify hypertension as early as possible. While there are no available data on how long early intervention delays the more severe renal and cardiac manifestations, it is clear that BP control in children with ADPKD is a clinical priority.
In contrast to the renal manifestations seen in ADPKD children and adults, the extrarenal manifestations of ADPKD commonly seen in adults are rarely seen in children.
2.2 Genetics - ADPKD
ADPKD is genetically heterogeneous with two known genes responsible for the vast majority of cases. PKD1 found on chromosome 16 is responsible for 80% of all ADPKD and PKD2, found on chromosome 4 is responsible for approximately 15% of ADPKD cases. Until recently approximately 5% percent of diagnosed ADPKD cases were genetically unresolved (GUR) [52]. Technical advances in whole genome sequencing and next-generation sequencing have spurred reexamination of cohorts of GUR cases. Additional genes are being identified that produce phenotypes that in many respects resemble ADPKD. The two most recent genes are GANAB [90], and DNAJB11 (DnaJ heat shock protein family (Hsp40) member B11) [91].
At least six genes (ALG8 (OMIM*608103), SEC61B (OMIM609214), LRP5 (OMIM *603506), SEC63 (*608648), PRKCSH (OMIM*177060), and most recently GANAB (*104160) [90] have been identified as causative genes for a distinct genetic entity, autosomal dominant polycystic liver disease (ADPLD). All six genes and DNAJB11 function in the ER as part of a quality control mechanism and mutations in any of these genes lead to reduced levels of PC1.
Genetic testing for the diagnosis of ADPKD is not generally necessary and technical challenges of analyzing PKD1 currently make it expensive. Cases where genetic testing may be helpful include clinical cases where there is no family history of PKD; reproductive counseling, and, for the identification of disease-free living related transplant donors.
Next-generation sequencing technologies offer increasingly cost-efficient diagnostic strategies which will ultimately reduce the cost. Prognostic assessment during the early stages of ADPKD will become increasingly important to identify patients with rapidly progressing disease who may be candidates for new clinical trials.
PKD1 is a large complex gene with 46 exons mapping to chromosome 16 (16p13.3). PKD1’s genomic structure has several characteristics that complicate its sequencing and consequently its use for molecular diagnostics:
-
it is a highly GC-rich gene with many CpG dinucleotides;
-
70% of the PKD1 gene, 5’ to exon 1 through exon 33, is duplicated six times proximally on chromosome 16. These duplications encode pseudogenes with high sequence homology (95% identity) to PKD1;
-
PKD1 contains a 2.5 kb polypyrimidine tract in intron 21 (the largest in the human genome) [92,93];
-
there is extreme allelic heterogeneity in both PKD1 and PKD2, with over 1500 different pathogenic mutations in PKD1, and 250 in PKD2;
-
algorithms for predicting the pathogenicity of DNA changes are lacking.
Genotype-phenotype correlations in ADPKD follow general patterns rather than absolute rules that begin with the well-recognized increased severity of PKD1 compared to PKD2. The average age of ESRD in PKD1 patients is 20 years earlier (53.4 versus 72.7) than PKD2 patients [52]. There is also a clear correlation between the type of mutation and disease severity. Truncating mutations (either frameshift, splicing, or nonsense) have an increased severity while non-truncating PKD1 mutations (missense, in-frame deletion/insertion) generally are associated with milder disease [94].
2.3 ADPKD Proteins
Polycystin 1 (PC1), the protein product of PKD1 is a large integral membrane protein of over 4000 amino acids. PC1 contains 11 transmembrane domains, an extensive extracellular domain with multiple predicted motifs, and a short intracellular C-terminal domain that regulates multiple signaling cascades [95]. PC1 is a putative receptor for an as yet unidentified ligand. The cytoplasmic carboxy-terminal coiled-coil domain of PC1 is known to interact with PC2; this interaction is required for PC1 maturation and trafficking to the cilia where PC1 and PC2 are thought to form a receptor-channel complex [96].
PC1 undergoes a series of post-translational cleavage events that alter the protein and its activity. PC1 undergoes a cleavage event at a juxtamembrane G protein-coupled receptor (GPCR) proteolytic site (GPS) motif generating a large N-terminal fragment (NTF) that remains noncovalently associated with the C-terminal fragment at the plasma membrane [97]. This cleavage seems to be a critical regulatory mechanism of PC1 maturation and trafficking [98]. Also, PC1 undergoes regulated cleavage of the intracellular C-terminal tail (CTT), resulting in the release of cytoplasmic fragments, which can translocate to the nucleus [99] and modulate signaling [100]. The CTT of PC1 has also been shown to translocate to the mitochondria [101]. Many if not most of the functions of PC1 remain unclear.
In contrast to PKD1, the PKD2 gene located on chromosome 4 (4q21) is much smaller, less complex with simpler features and structure [102]. PKD2 has 15 exons, spanning 68 kb of genomic sequence, encoding a 2904 bp open reading frame transcript [102]. Polycystin 2 (PC2), the product of the PKD2 gene, is a member of the transient receptor potential (TRP) family of nonselective cation channels [103]. Polycystin-2 is a calcium-permeable channel with six transmembranes (TM) spanning elements, and both the amino and carboxy termini face the cytoplasmic compartment. Interaction with PC1 is required for full-length PC2 to leave the ER and reach the cilia [96,104].
3. Pathophysiology of Cyst Formation
The process of cyst formation and progressive growth is a multifaceted, dynamic process with multiple elements all contributing to the process, but never acting autonomously. The PKD1 and PKHD1 genes demonstrate a genetic interaction [105] and the proteins FPC and PC2 have been shown to directly interact [104,113,114] [#. PC1, PC2, and PKHD1 are expressed in several cellular compartments and form multimeric protein complexes that modulate signaling pathways, which control essential cellular functions. This provides a molecular basis for the similar phenotypic expression of childhood ARPKD and ADPKD. For the clinician, it implies that therapies effective in either cystic disease may be effective in the other.
These proteins are localized to apical and basolateral plasma membranes, desmosomes, focal adhesions, basal bodies, and primary cilium. Except for PC2, there is little evidence to indicates the actual function of the PKD proteins PC1 or FPC or how these protein complexes may communicate with each other.
The rapid development of sophisticated techniques in molecular biology and increased specificity in producing targeted gene mutations has led to the development of new orthologous mouse models with conditional and hypomorphic mutations in Pkd1, Pkd2, and Pkhd1[106]. In combination with improved next-generation sequencing techniques, the ability to create specific targeted mutations has allowed animal models with mutations identified in PKD patients to be generated to test a mechanistic hypothesis. Such models have revealed unanticipated results regarding the development and progression of PKD. These new insights will lead to novel methods and novel compounds for therapeutic intervention for PKD.
A remarkable feature of PKD is the variable phenotypic disease expression in both ADPKD and ARPKD. Gaining a better understanding of disease progression and identifying determinants of disease severity will provide multiple benefits. It will identify new therapeutic avenues where adjustments of one or more of determinants of severity can be targeted to reduce disease progression. Further, it will serve to identify candidates most likely to benefit from a proposed clinical trial. Finally, it will identify patients that are unlikely to benefit from a specific therapy thereby preventing unnecessary and possibly harmful side-effects of a clinical trial.
3.1 Molecular Pathophysiology
Factors Affecting Progression or Severity. Variants in PKD alleles, as well as complex transcriptional events, play a key role in modifying progression and severity of childhood PKD. Recent discoveries of diverse transcriptional patterns and allelic variants including: hypomorphic or incompletely penetrant alleles [107,108]; homozygosity of PKD1 or PKD2 [108]; compound heterozygosity [8]; germline mosaicism [109]; digenic inheritance or trans-heterozygosity [110]; co-inheritance of mutations in either PKD1 or PKD2 and an additional PKD-causing gene such as PKHD1 [105], HNF1B [14,111] or the tuberous sclerosis 2 (TSC2) gene [112]; all demonstrate that ADPKD and ARPKD have complex inheritance patterns which can contribute to disease severity.
In addition to the molecular transcriptional events listed above, other molecular events that highlight interactions between the PKD genes and proteins have been discovered that influence the severity and rate of cyst formation and total kidney growth as well.
The genetic combination of five murine cystic disease models including orthologous Pkd1, Pkd2, Pkhd1, and two orthologous ADPLD genes, Prkcsh and Sec63 allowed the creation of different combinations of mutant alleles to characterize the functional relationships between these genes [113]. These combinations yielded unanticipated relationships and surprising results. These crosses demonstrated that:
-
PC1 dosage modifies the severity of both ADPKD and ARPKD
-
the threshold level of PC1 necessary for normal tubular morphology varies by nephron segment with collecting ducts being most sensitive;
-
overexpression of Pkd1 can rescue a mutant Pkhd1 [113];
-
Pkhd1 and Pkd1 interact at the gene level;
-
the overexpression PC1 can cause cysts.
These studies demonstrated that the rate of cyst growth is a regulated trait, which can be adjusted by changes in levels of functional PC1 [114].These data support a threshold or dosage model of PKD. According to this model, cyst initiation and cystic expansion are governed by Pkd1 gene dosage [113,114,115]. Every cell or nephron has a threshold level of Pkd1 that is necessary to maintain normal tubular size and maintain cells in a non-proliferative state. When the level of functional PC1 falls below the cystogenic threshold, proliferation and cyst formation follow.
In a similar experiment tissue-specific, conditional and inducible mouse models that deleted structural ciliary genes (Kif3a and Ift20) and PKD genes (Pkd1, Pkd2, and Pkhd1), in a spatiotemporal manner,alone or in combination were examined to determine the functional roles of PKD1 and PKD2 in cilia. The various combinations revealed that:
-
loss of cilia causes less cyst growth than the loss of PC1 or PC2;
-
simultaneous loss of either PC and either the cilia structural proteins Kif3a or Ift20 resulted in greater reduction in cyst formation than that seen when either PC was inactivated; and
-
the length of time that intact cilia existed after the loss of PC’s increased disease severity [116,117]. These data suggest that the existence of a cilia-dependent, cyst-promoting (proliferation promoting) pathway exists which is inhibited by a normal PC1/PC2 complex on the cilia [116,117,118].
The two-hit theory of cyst development originated from the observation that ADPKD is a molecularly recessive disease and the cystic lesions are focal. All cells in the body have the same mutation, but estimates assert that only 5 to 10% of nephrons become cystic. The two-hit theory states that a second somatic mutation is required for cyst formation to occur. Substantial evidence exists to support this theory [119,120,121]. However, other factors have been shown to influence disease progression and severity [122]. These include:
-
the developmental timing of PKD1 inactivation [123,124,125,126];
-
reduction in functional PC1 dosage; [107,108,113,114]. The reduction of PKD1 dosage below 50% is sufficient to cause cystic lesions;
-
renal cells cellular and nephron segments show differences in sensitivity to PC1 dosage and have different requirements for the amount of PC1 needed for normal development/function [114];
-
The principal cells of the collecting duct appear to be the most sensitive [108];
-
the effect of one cyst on neighboring cells and nephrons creating a toxic environment or a “snowball effect” leading to cyst development in neighboring nephrons [127];
Therapies targeting genetic mechanisms in ADPKD have inherent limitations. As a result, most experimental therapies at present are aimed at delaying the growth of the cysts and associated interstitial inflammation and fibrosis by targeting cystic tubular epithelial cell proliferation and fluid secretion by the cystic epithelium.
3.2 Targeting the Cystic Phenotype
The specific mechanisms that accelerate cyst formation and progressive enlargement are poorly understood. However; it is clear that essential phenotypic changes are required for a normal absorptive, non-proliferating tubular epithelial cell to become a cystic epithelial cell. These changes include the proliferation of cells which line expanding cysts, fluid secretion into cysts that have lost their connection to the nephron and remodeling of the extracellular matrix surrounding the expanding tubules. In PKD, normal renal epithelial cells change from mature, differentiated, non-proliferative, absorptive cells to partially de-differentiated secretory cystic cells characterized by specific polarity defects and increased rates of proliferation.
Identification of signaling molecules and signaling pathways associated with PKD define the “cystic phenotype.” The precise mechanisms by which mutated PKD proteins alter signaling molecules and integrate signaling pathways are not fully understood, and there is little known about the actual function of the proteins PC1 and FPC. Despite this gap in knowledge, key pathogenic features of both ARPKD and ADPKD have been identified and form the basis of current therapeutic interventions. These features include but are not limited to:
-
Altered intracellular cAMP levels coupled with decreased intracellular calcium;
-
Abnormalities in expression, localization, and activity of the epidermal growth factor (EGFR) – a family of receptors and ligands (EGFR axis);
-
Abnormal activity of cSrc, a critical mediator of cross-talk between the EGFR axis and G-protein-cAMP pathways which is required for STAT3 activation by a PC1 tail;
-
Alterations in cell-cell adhesion, and cell-matrix interactions;
-
Specific interstitial-tubular interactions which lead to chronic Inflammation and progressive fibrosis.
Clinical trials to date have been largely based on targeting these aberrant cellular signaling elements which define the cystic phenotype. Figure 1 is a cartoon that includes the major aberrant signaling pathways that comprise the cystic phenotype.
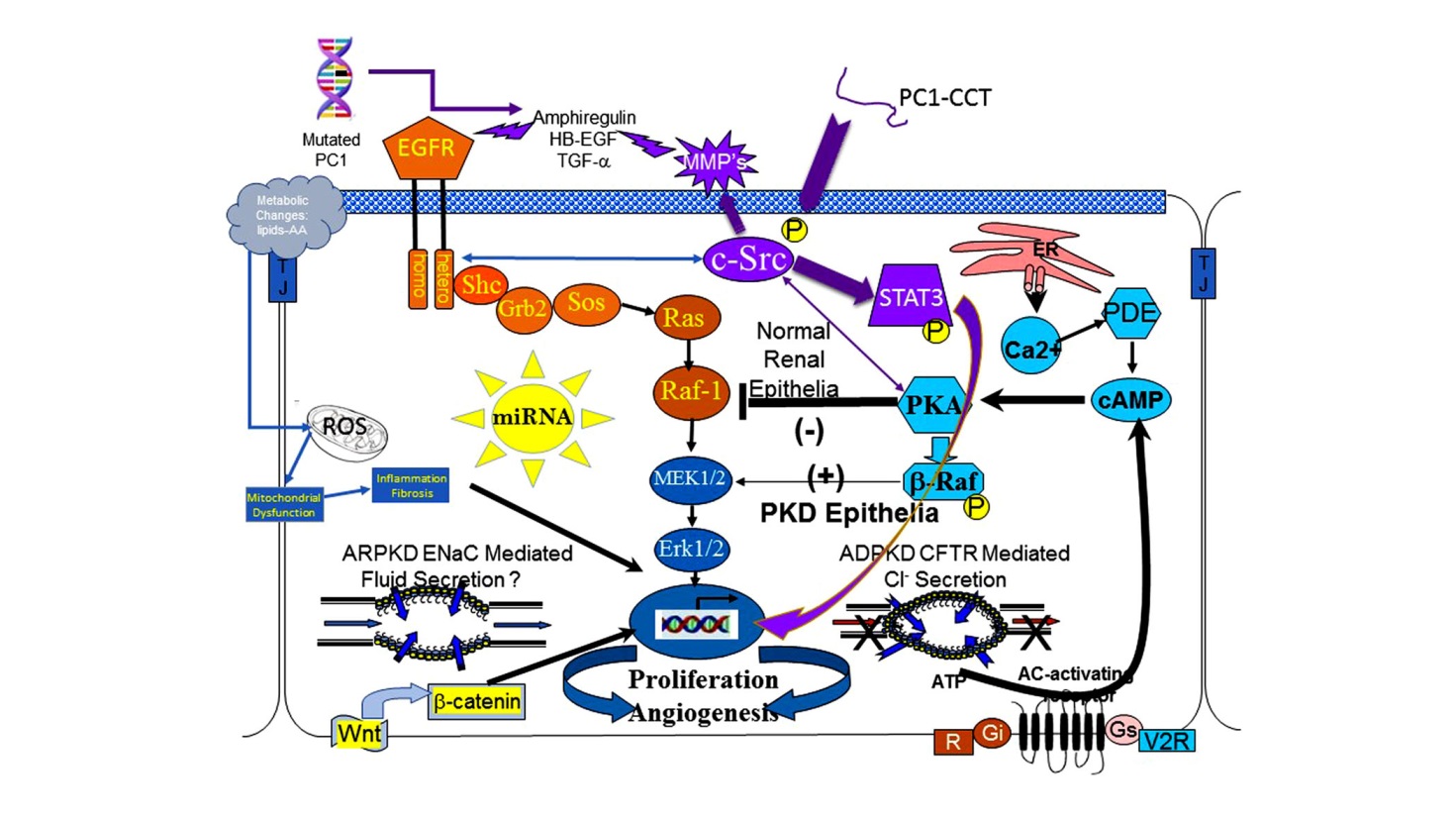
Figure 1 The cystic cellular phenotype. This cartoon is an abridged composite of the abnormal signal transduction pathways reported to be active in PKD. Two main conduits that lead to unchecked proliferation are: 1) the EGFR axis (orange); and 2) a G-protein axis (aqua blue) that leads to increased cAMP and a switch in the phenotypic response of renal epithelia to cAMP. The pathway suggest the following possible scenario: In ARPKD, an apical EGFR becomes activated resulting in reciprocal phosphorylation of the nonreceptor tyrosine kinase cSrc leading to increased cAMP-mediated, and EGFR driven proliferation; and in ADPKD (purple) a mutated PC1 leads to increase amphiregulin, activating EGFR, resulting in increased cSrc phosphorylation; in both ARPKD and ADPKD, cSrc activation alters the cellular response of cAMP resulting in increased proliferation and secretion; in addition, the cytoplasmic tail, PC1-p30, is overexpressed. PC1-p30 interaction with Src causes Src-dependent activation of STAT3 by tyrosine phosphorylation. Increased activity of EGFR and increased cAMP amplify the activation of the Src-STAT3 pathway. Finally, aberrant miRNA expression can impact any pathway and may effect more than one at a time.
cAMP-mediated proliferation and secretion. The cAMP-dependent pathway, or the adenylyl cyclase pathway, is a G-protein-coupled receptor signaling pathway that is abnormally regulated and aberrantly active in PKD. In normal renal epithelia increased intracellular cyclic AMP (cAMP) levels result in reduced epithelial proliferation. However, in cystic renal epithelia, a low intracellular calcium level, (theoretically due to a defective PC1/PC2 calcium channel) and a cSrc dependent phosphorylation of b-Raf, allows the cell to bypass Raf-1 and increase ERK phosphorylation to stimulate cell proliferation in the presence of increased cAMP levels [128,129]. Increased cAMP in a cell with an active MAPK pathway also changes the cell from an absorptive to a secretory cell which contributes significantly to progressive cyst enlargement in ADPKD [130].
Therapeutically, increased cAMP is reduced by vasopressin R2 receptor antagonists such as tolvaptan (Samsca or Jinarc®) or somatostatin(and its long-acting analogs such as lanreotide that inhibit adenylate cyclase thereby reducing cAMP levels [131,132].
Tolvaptan has undergone extensive human clinical trials. Tolvaptan slows the increase in total kidney volume and loss of renal function [133,134,135]. Jinarc® has been approved in Europe, Japan, Canada, Australia, and South Korea for the treatment of adults with ADPKD. Tolvaptan has recently been approved in the USA under the trade name JYNARQUE®. Due to concerns regarding potentially serious adverse liver events, JYNARQUE is only available in the USA through a restricted distribution program called the JYNARQUE Risk Evaluation and Mitigation Strategy which is designed to closely monitor patients for liver or other adverse events.Details can be found at: (www.fda.gov/safety) and (https://www.jynarque.com).
A single report of the use of tolvaptan in severe neonatal ADPKD was found to resolve hyponatremia and stop the growth of the kidneys when administered betweenPN30 to 12 months of age [136]. The medical team instituted a strict monitoring program that included frequent blood pressure, urine, and electrolytes and hepatic function testing [137]. Despite this successful use of tolvaptan in a child, Ttolvaptan is not currently recommended for use in children due to the necessity to closely monitor fluid intake in the pediatric population. Never the less, a Phase III clinical trail of tolvaptan in children between age 4 and 17 years with ADPKD is under way in Belgium, Italy, Germany and the United Kingdom (NCT02964273). A clinical trial of Tolvaptan in children and adolescents with euvolemic and hypervolemic hyponatremia will provide additional data on the safety, pharmacokinetics and fluid balance issues with tolvaptan use in a pediatric population (Clinical Trials.gov Identifier: NCT02442674).
Palladia Sciences is testing a drug called Lixivaptan that the company believes provides the same benefits of Ttolvaptan in ADPKD without the potential liver issues associated with Tolvaptan use (ELSA study NCT 03487913). In addition to the potential problems on liver function with tolvaptan therapy, the unanticipated finding of increased shedding of HB-EGF in the urine of patients treated with tolvaptan may be problematic especially with lengthy use due to the known role of EGFR in fibrosis [137]. If this data is confirmed, it suggests the addition of a tyrosine kinase inhibitor (TKI) or multi-TKI to the tolvaptan treatment may improve clinical results.
EGFR (ErbB) axis. The epidermal growth factor receptor (EGFR) axis consist of the epidermal growth factor/ErbB family of receptors and ligands. A large body of evidence implicates one or more members of the ErbB receptor family in renal cystic epithelial proliferation, including the epidermal growth factor receptor (EGFR), as well as the related receptors, ErbB2 and ErbB4 [138,139,140].
In both human ADPKD and ARPKD, and every rodent model of PKD published to date, cystic kidneys display characteristic alterations in EGFR-axis expression. Inhibition or reduction of EGFR activity through genetic manipulation [141] or administration of a tyrosine kinase inhibitor [142,143], reduces cyst formation and progressive enlargement in murine cystic models.
Recent studies have provided a direct link between PC1 and PC2 proteins and the EGFR axis. These studies have revealed that EGF reduces the threshold of PKD2 activation by releasing it from PIP(2)-mediated inhibition [144] and that mutated PC1 results in increased expression of amphiregulin and increased proliferation [145].
Microarray profiling in normal human and cystic PKD1 kidney cells has demonstrated that the EGF/ErbB family receptor (ErbB4) is a major driver of cyst growth in ADPKD and ErbB4 levels also serve as a potential biomarker for disease progression [146]. This increased expression of ErbB4 in vivo in both human ADPKD and Pkd1 cystic kidneys was associated with a single microRNA (miRNA), mir-193b-3p [146].
cSrc. c-Src is a critical intermediate that integrates signals between two major proliferative pathways in cystic epithelial cells, the EGFR axis, and cyclic AMP pathway [147] (see Figure 1). Also, a proteolytic fragment of the PC1 cytoplasmic tail, PC1-p30, interacts with cSrc, to activate STAT3 stimulating further increases in proliferation [148,149,150]. Src activation of STAT 3 is amplified by both the increases in activity of the EGFR axis and by increased cAMP levels resulting in a self-perpetuating cycle that accelerates proliferation [150].
These experimental results would predict a pathologic pathway where a mutation in PC1 increases amphiregulin expression, resulting in activation of the EGFR axis (EGFR or ErbB4) and reciprocal activation of Src. In concert with increased cAMP levels and cleavage of the PC1 tail, cSrc assimilates and amplifies input from the EGFR axis, and cAMP signaling pathways to activate STAT3 [150].
In a clinical trial of Bosutinib, a cSrc inhibitor (ClinicalTrials.gov Identifier NCT01233869) was very effective in reducing total kidney volume in ADPKD patients. However, in this limited clinical trial, bosutinib did not affect changes in renal function [151].
mTOR. The mammalian target of rapamycin (mTOR) pathway integrates signals from growth factors (including EGFR), G-protein coupled receptors (which generate cAMP), cellular energy levels, nutrient status and stress conditions to stimulate protein synthesis and cell growth through activation (phosphorylation) of S6K1 and eIF4E [152,153]. In human ADPKD, ARPKD and a variety of animal models, cyst-lining epithelium reveals an increased activity of mTOR [153,154,155,156]. The mTOR inhibitors rapamycin and everolimus have been tested in human clinical trials and were found to be ineffective in slowing total kidney volume or the progressive loss of renal function [157,158]. It has been hypothesized that effective renal concentration of mTOR inhibitors was not achieved and thereby responsible for the disappointing clinical results. Based on the finding that cystic cells express folate receptors Shillingford et al. conjugated folate to rapamycin. The effectiveness of mTOR inhibition was dramatically improved, and target specificity was increased in a Pkd1 animal model when rapamycin was conjugated with folate [159]. To date, no clinical trials have been reported utilizing this new approach to mTOR inhibition in PKD.
3.3 Emerging Therapies
Tesevatinib. Tesevatinib (TSV) previously known as KD-019, is a unique multi-kinase inhibitor that targets EGFR, c-Src and KDR. TSV was effective in slowing the proliferation of renal epithelia and cyst growth, slowing progression of fibrosis and slowing the loss of renal function in the BPK and orthologous PCK rat models of ARPKD [160].
TSV is currently being tested in two adult Phase II-III clinical trials in ADPKD [ClinicalTrials.gov Identifier NCT01559363 and NCT03203642. TSV is also the first specific disease-targeted therapy to receive FDA approval for a clinical trial in pediatric ARPKD [ClinicalTrials.gov Identifier NCT03096080]. All three clinical trials are in progress.
20 – HETE. Metabolic changes are known to occur during the development and progression of ARPKD and ADPKD. Bioactive lipids such as 20-HETE, a P450-produced bioactive lipid metabolite of arachidonic acid was shown to correlate with eGFR and TKV in ADPKD patients [161]. Three studies demonstrated the effectiveness of 20 HETE inhibition an animal models of PKD [162,163,164] indicated that inhibitors of 20-HETE production may have potential use as a therapeutic agent for slowing PKD progression and as a non-invasive biomarker to monitor disease desease progression.
Bardoxolone Methyl or Bard (Reata Pharmaceuticals). Triterpenoids are used for medicinal purposes in many Asian countries because of their anti-inflammatory, antioxidant, antiproliferative, anticancer, and anticarcinogenic properties. Bardoxolone methyl is an experimental and orally-bioavailable semi-synthetictriterpenoid that in pre-clinical studies acts as an activator of theNrf2pathway andinhibitor of the transcription factor NF-kB (Reata Pharmaceuticals). Nrf2regulates the expression of antioxidant proteins that protect against oxidative damage triggered by injury and inflammation, and NF-kb is a pro-inflammatory transcription factor. Inflammation and oxidative damage are both implicated in all forms of CKD including PKD, regardless of initiating cause [165].
Twenty-seven clinical trials have been completed or are in progress utilizing BARD to target cancer and chronic renal diseases. A Phase II multi-center clinical trial (ClinicalTrials.gov Identifier: NCT00664027) of BARD in patients with moderate to severe CKD and type 2 diabetes reported that eGFR increased as much as 10 ml/min/1.73 m² in patients treated with bardoxolone methyl for 24 months [166]. To date, no therapy targeting ARPKD or ADPKD has shown to improved eGFR. At best, therapies have been shown to reduce the rate of functional decline.
Recruitment is underway for a Phase II Trial of the Safety and Efficacy of Bardoxolone Methyl in Patients With Rare Chronic Kidney Diseases associated with type 1 diabetes (T1D), IgA nephropathy (IgAN), focal segmental glomerulosclerosis (FSGS), and autosomal dominant polycystic kidney disease (ADPKD) (ClinicalTrials.gov Identifier: NCT03366337). The estimated completion date is August 2019.An improved eGFR if found following Bard treatment of PKD would be a remarkable finding.
MicroRNAs.Given the complexity of the ADPKD genes and the multiple interactions discussed above, therapeutic interventions are not likely in the near future with the possible exception of miRNA inhibition. miRNAs are small, non-coding RNAs with a strictly regulated biogenesis. This is combined with an extremely flexible and sophisticated regulatory function of gene expression at the post-transcriptional level. This allows the simultaneous targeting of multiple proteins involved in different crucial biological pathways of specific cell types and tissues. miRNAs are integral to the normal development and physiology of the human kidney. They have also been shown to be central to the pathophysiology of many human disease states including renal diseases. As mentioned above, a recent study in human ADPKD has linked a single miRNA to the progression of human ADPKD [146] and more recently a unique interstitial miRNA signature was found to play a role in the pathogenesis of renal fibrosis in an orthologous animal model of ADPKD [167]. Recent studies have also identified miRNAs that play a role in the pathogenesis of PKD. Deletion or inhibition of the miR-17-92 cluster in four orthologous animal models of PKD, and cell cultures from multiple human cysts result in reduced epithelial proliferation and cyst growth [168,169,170].
3.4 Non-Specific Therapies Targeting PKD
Hypertension.In ADPKD, hypertension is associated with increased TKV, activation of the renin-angiotensin-aldosterone system (RAS) and progressive loss of renal function. Therefore, the identification and treatment of hypertension is essential in slowing the progression to ESRD in ADPKD.
The NIH sponsored HALT/PKD studies failed to demonstrate any benefit effect of adding an ARB (telmisartan) to an ACEi (lisinopril) on reducing disease progression regardless of level of renal function in adult ADPKD patients. However, in younger patients with moderately preserved renal function, the reduction of blood pressure in children and young adults (age 15 to 49) a lower target level with an ACEi significantly slowed the increase in TKV and decreased urinary albumin excretion and left ventricular mass index but did not slow the decline in renal function. estimated glomerular filtration rate [68,171].These data support the premise that targeting lower BP goals in ADPKD patients with RAS blockade may have long term beneficial effects in preserving renal function, and may be particularly beneficial to children and young adults.
Statins Therapy. The cholesterol-lowering drugs, 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase inhibitors, collectively called statins, have anti-proliferative, anti-inflammatory and anti-oxidant effects independent of cholesterol lowering effects [172,173].
A randomized double-blind placebo controlled pPhase III clinical trial in pediatric and adolescence patients with ADPKD demonstrated that statin therapy significantly slowed increases in TKV and reduced the decline of renal function over a three year period [174]. To date, these results have not been replicated in adults with ADPKD but a clinical trial still recruiting patients is designed to examine this issue (ClinicalTrials.gov Identifier: NCT03273413).
Dietary Modification.
Water consumption:Suppression of serum AVP by increasing free water intake would be predicted to decrease intracellular cAMP concentrations in collecting duct cells and thereby reduce tubular epithelial cell proliferation and cyst growth. A number of small observational trials have looked at the effect of a short-term increased in water consumption on the progression of PKD. Thus far the data has been intriguing not been persuasive enough to support recommending the use of increased water consumption to slow progression of disease. However, clinical trials are underway to examine the effect of increased water consumption to achieve a urine osmolality < 270 mOsmo/kg or urine specific gravity ≤ 1.010 (ClinicalTrials.gov Identifier: NCT02933268). One such study in the USA isstill recruiting patients (ClinicalTrials.gov Identifier: NCT03102632) as is a study in Australia (Trial registration number ANZCTR12614001216606 [173].
Caloric Restriction and Other Dietary Recommendations: A recent study in mouse models of PKD examined the effect of caloric restriction on disease progression [175]. The study revealed a mild to moderate (10%-40%) reduction of food slowed disease progression in a dose-dependent manner. These findings have ignited an interest in examining the effect of caloric restriction on ADPKD progression. A clinical trial to assess the effect of “Daily Caloric Restriction and Intermittent Fasting in Overweight and Obese Adults with ADPKD (ClinicalTrials.gov Identifier: NCT03342742) is underway.
4. Conclusions
The diagnosis of childhood PKD is no longer the terminal diagnosis as it once was. For children with ARPKD advances in neonatal critical care, control of hypertension and hepatic complications, and renal/hepatic transplantation have allowed many to survive into adulthood. Insights into the development and treatment of PH are preventing lethal complications of hepatobiliary disease and provide a better quality of life. Renal transplantation and dual organ transplantation provide an opportunity for these children to live a near normal life [22].
Pre-implantation genetic diagnosis (PGD) and in vitro fertilization (PGD-IVF) holds the possibility of eliminating ARPKD from families who can afford the procedure and who have no religious/ethical contraindications [51]. PGD for ADPKD is becoming more widely available [176] and PGD-IVF for ADPKD is being examined under clinical trial in China (NCT02948179).
As clinical trials to date have shown, targeting a single molecule of an aberrant pathway has not been effective in preventing the loss of renal function as originally hoped. However, even as the targeted therapeutic strategies develop more specificity, they will always need to target (a) cell proliferation; (b) cAMP-mediated proliferation and secretion; and (c) interstitial fibrosis. Proliferation is an essential component that must be targeted and controlled for any therapy to be effective. How much and for how long remains are questions currently under investigation.
In the short term, the most promising therapies will likely target key signaling intermediates that appear to integrate multiple pathways, such as Src, (see Figure 1) [147,150] and/or utilize multiple compounds to target multiple pathways simultaneously or a single compound that targets multiple pathways. A single compound like TSV that targets multiple pathways [177,178], offers great promise. Future rational therapies will also require a knowledge of the extent of disease, as well as the rate of progression, and will likely involve different compounds that will vary at specific stages of disease progression.
There are additional essential factors to consider in developing therapies for childhood PKD. Therapies will become increasingly focused on treating PKD in early childhood where they are likely to provide the maximum long-term benefit. Therapies that target abnormal signaling pathways will need to be carefully tailored, so that pathway activity is reduced to normal basal levels rather than eliminating the signal completely. Promising compounds will be modified to direct the molecule to the site of disease, making such therapies highly specific with low levels of toxicity. This specificity will speed the development of protocols for the ethical treatment of children with PKD so early intervention will provide maximal quality of life. Epigenetic and dietary factors that slow or accelerate the progression of PKD will be discovered, and adherence or avoidance of such factors may slow the rate of progression and may eliminate the need for pharmacological intervention or renal replacement therapy for some.
The importance of gene dosage, modifier genes and somatic mutations in the clinical course of a disease in an individual patient provides a compelling rationale for personalized medicine in PKD. Such therapies will modulate functional gene dosage and base therapy not only on the individual genotypes but will account for the response of the kidney and liver to the therapy itself and any unanticipated response of the kidney or the patient to the therapy.
The progress made in understanding the pathophysiology of PKD has been remarkable and despite the progress, work remains cClinical trials for children with PKD are underway: One for children with ADPKD [Tolvaptan [NCT02964273 Belgium and Italy] and one recently approved for the use of TSV in ARPKD (ClinicalTrials.gov Identifier NCT03096080). A list of current and completed clinical trials can be found at http://clinicaltrials.gov/.
Acknowledgments
This work was supported, in part, by the Amy P. Goldman Charitable Trust; the Ellsworth Family Fund; and the Children’s Research Institute at the Children’s Hospital of Wisconsin.
Author Contributions
Author Contributions: AP, WES, and EDA all contributed substantially to the organization and drafting of the review and all authors made critical revisions related to the intellectual content of the manuscript, and approved the final version of the article to be published.
Competing Interests
The authors declare that they have no conflicts of interest concerning research, authorship and publication of this manuscript.
References
- Zerres K, Mucher G, Becker J, Steinkamm C, Rudnik-Schoneborn S, Heikkila P, et al. Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical experience, and fetal morphology. Am J Med Genet. 1998; 76: 137-144. [CrossRef]
- Gunay-Aygun M, Avner ED, Bacallao RL, Choyke PL, Flynn JT, Germino GG, et al. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis: summary statement of a first National Institutes of Health/Office of Rare Diseases conference. J Pediatr. 2006; 149: 159-164. [CrossRef]
- Dell KM. The spectrum of polycystic kidney disease in children. Adv Chronic Kidney Dis. 2011; 18: 339-347. [CrossRef]
- Gunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman Gerstein M, Piwnica-Worms K, Choyke P, et al. Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology. 2013; 144: 112-121. [CrossRef]
- Sweeney WE, Jr., Avner ED. Pathophysiology of childhood polycystic kidney diseases: new insights into disease-specific therapy. Pediatr Res. 2014; 75: 148-157. [CrossRef]
- Zerres K, Rudnik-Schoneborn S, Steinkamm C, Becker J, Mucher G. Autosomal recessive polycystic kidney disease. J Mol Med. 1998; 76: 303-309. [CrossRef]
- Bergmann C, Zerres K. Early manifestations of polycystic kidney disease. Lancet. 2007; 369: 2157. [CrossRef]
- Rossetti S, Harris PC. Genotype-phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease. J Am Soc Nephrol. 2007; 18: 1374-1380. [CrossRef]
- Guay-Woodford LM, Bissler JJ, Braun MC, Bockenhauer D, Cadnapaphornchai MA, Dell KM, et al. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. J Pediatr. 2014; 165: 611-617. [CrossRef]
- Sweeney WE, Jr., Gunay-Aygun M, Patil A, Avner ED. Childhood Polycystic Kidney Disease. In: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Emma F, Goldstein S, editors. Pediatric Nephrology; 2016. p. 1-58. [CrossRef]
- Guay-Woodford LM. Autosomal recessive polycystic kidney disease: the prototype of the hepato-renal fibrocystic diseases. J Pediatr Genet. 2014; 3: 89-101.
- Bergmann C, Senderek J, Windelen E, Kupper F, Middeldorf I, Schneider F, et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 2005; 67: 829-848. [CrossRef]
- Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics. 2014; 134: e833-e845. [CrossRef]
- Bergmann C. ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocopies. Pediatr Nephrol. 2014.
- Kaariainen H, Koskimies O, Norio R. Dominant and recessive polycystic kidney disease in children: evaluation of clinical features and laboratory data. Pediatr Nephrol. 1988; 2: 296-302. [CrossRef]
- Zerres K, Rudnik-Schoneborn S, Deget F, Holtkamp U, Brodehl J, Geisert J, et al. Autosomal recessive polycystic kidney disease in 115 children: clinical presentation, course and influence of gender. Arbeitsgemeinschaft fur Padiatrische, Nephrologie. Acta Paediatr. 1996; 85: 437-445. [CrossRef]
- Lieberman E, Salinas-Madrigal L, Gwinn JL, Brennan LP, Fine RN, Landing BH. Infantile polycystic disease of the kidneys and liver: clinical, pathological and radiological correlations and comparison with congenital hepatic fibrosis. Medicine. 1971; 50: 277-318. [CrossRef]
- Gagnadoux MF, Habib R, Levy M, Brunelle F, Broyer M. Cystic renal diseases in children. Adv Nephrol Necker Hosp. 1989; 18: 33-57.
- Kaplan BS, Fay J, Shah V, Dillon MJ, Barratt TM. Autosomal recessive polycystic kidney disease. Pediatr Nephrol. 1989; 3: 43-49. [CrossRef]
- Gunay-Aygun M, Gahl WA, Heller T. Congenital Hepatic Fibrosis Overview Seattle (WA): University of Washington; 2008. Available from: http://www.ncbi.nlm.gov/books/NBK2701.
- Turkbey B, Ocak I, Daryanani K, Font-Montgomery E, Lukose L, Bryant J, et al. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis (ARPKD/CHF). Pediatr Radiol. 2009; 39: 100-111. [CrossRef]
- Telega G, Cronin D, Avner ED. New approaches to the autosomal recessive polycystic kidney disease patient with dual kidney-liver complications. Pediatr Transplant. 2013; 17: 328-335. [CrossRef]
- Adeva M, El-Youssef M, Rossetti S, Kamath PS, Kubly V, Consugar MB, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore). 2006; 85: 1-21. [CrossRef]
- Cole BR, Conley SB, Stapleton FB. Polycystic kidney disease in the first year of life. J Pediatr. 1987; 111: 693-699. [CrossRef]
- Sweeney WE, Jr., Avner ED. Diagnosis and management of childhood polycystic kidney disease. Pediatr Nephrol. 2011; 26: 675-692. [CrossRef]
- Sweeney WE, Avner ED. Polycystic Kidney Disease, Autosomal Recessive. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R). Seattle (WA)1993.
- Harris T, Sandford R, de Coninck B, Devuyst O, Drenth J, Ecder T, et al. European ADPKD Forum multidisciplinary position statement on autosomal dominant polycystic kidney disease care. Nephrol Dial Transplant. 2018; 33: 563-573. [CrossRef]
- Guay-Woodford LM, Henske E, Igarashi P, Perrone RD, Reed-Gitomer B, Somlo S, et al. Filling the holes in cystic kidney disease research. Clin J Am Soc Nephrol. 2014; 9: 1799-1801. [CrossRef]
- Bullo M, Tschumi S, Bucher BS, Bianchetti MG, Simonetti GD. Pregnancy outcome following exposure to angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists: a systematic review. Hypertension. 2012; 60: 444-450. [CrossRef]
- Guay-Woodford. Autosomal recessive polycystic kidney disease: clinical and genetic profiles. In: Watson ML, Torres VH, editors. Polycystic Kidney Disease. New York: Oxford University Press; 1996. p. 237-266.
- Gunay-Aygun M. Liver and kidney disease in ciliopathies. Am J Med Genet C Semin Med Genet. 2009; 151C: 296-306. [CrossRef]
- Lu H, Galeano MCR, Ott E, Kaeslin G, Kausalya PJ, Kramer C, et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat Genet. 2017; 49: 1025-1034. [CrossRef]
- Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002; 70: 1305-1317. [CrossRef]
- Boddu R, Yang C, O'Connor AK, Hendrickson RC, Boone B, Cui X, et al. Intragenic motifs regulate the transcriptional complexity of Pkhd1/PKHD1. J Mol Med (Berl). 2014; 92: 1045-1056. [CrossRef]
- Bergmann C, Senderek J, Kupper F, Schneider F, Dornia C, Windelen E, et al. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat. 2004; 23: 453-463. [CrossRef]
- Gunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman M, Graf J, Bryant JC, et al. Correlation of kidney function, volume and imaging findings, and PKHD1 mutations in 73 patients with autosomal recessive polycystic kidney disease. Clin J Am Soc Nephrol. 2010; 5: 972-984. [CrossRef]
- Bergmann C, Senderek J, Sedlacek B, Pegiazoglou I, Puglia P, Eggermann T, et al. Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1). J Am Soc Nephrol. 2003; 14: 76-89. [CrossRef]
- Rossetti S, Torra R, Coto E, Consugar M, Kubly V, Malaga S, et al. A complete mutation screen of PKHD1 in autosomal-recessive polycystic kidney disease (ARPKD) pedigrees. Kidney Int. 2003; 64: 391-403. [CrossRef]
- Bergmann C, Senderek J, Windelen E, Kupper F, Middeldorf I, Schneider F, et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 2005; 67: 829-848. [CrossRef]
- Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002; 30: 259-269. [CrossRef]
- Bergmann C, Zerres K. Polycystic Kidney Disease:ADPKD and ARPKD. In: Geary DF, Schaefer F, editors. Comprehensive Pediatric Nephrology. Philadelphia: Mosby (Elsevier); 2008. p. 155-178. [CrossRef]
- Sharp AM, Messiaen LM, Page G, Antignac C, Gubler MC, Onuchic LF, et al. Comprehensive genomic analysis of PKHD1 mutations in ARPKD cohorts. J Med Genet. 2005; 42: 336-349. [CrossRef]
- Furu L, Onuchic LF, Gharavi A, Hou X, Esquivel EL, Nagasawa Y, et al. Milder presentation of recessive polycystic kidney disease requires presence of amino acid substitution mutations. J Am Soc Nephrol. 2003; 14: 2004-2014. [CrossRef]
- Gunay-Aygun M, Tuchman M, Font-Montgomery E, Lukose L, Edwards H, Garcia A, et al. PKHD1 sequence variations in 78 children and adults with autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Mol Genet Metab. 2010; 99: 160-173. [CrossRef]
- Szabo T, Orosz P, Balogh E, Javorszky E, Mattyus I, Bereczki C, et al. Comprehensive genetic testing in children with a clinical diagnosis of ARPKD identifies phenocopies. Pediatr Nephrol. 2018. [CrossRef]
- Frank V, Zerres K, Bergmann C. Transcriptional complexity in autosomal recessive polycystic kidney disease. Clin J Am Soc Nephrol. 2014; 9: 1729-1736. [CrossRef]
- Nagasawa Y, Matthiesen S, Onuchic LF, Hou X, Bergmann C, Esquivel E, et al. Identification and characterization of Pkhd1, the mouse orthologue of the human ARPKD gene. J Am Soc Nephrol. 2002; 13: 2246-2258. [CrossRef]
- Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, et al. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol. 2007; 27: 3241-3252. [CrossRef]
- Kaimori JY, Nagasawa Y, Menezes LF, Garcia-Gonzalez MA, Deng J, Imai E, et al. Polyductin undergoes notch-like processing and regulated release from primary cilia. Hum Mol Genet. 2007; 16: 942-956. [CrossRef]
- Tabrez SS, Sharma RD, Jain V, Siddiqui AA, Mukhopadhyay A. Differential alternative splicing coupled to nonsense-mediated decay of mRNA ensures dietary restriction-induced longevity. Nat Commun. 2017; 8: 306. [CrossRef]
- Lau EC, Janson MM, Roesler MR, Avner ED, Strawn EY, Bick DP. Birth of a healthy infant following preimplantation PKHD1 haplotyping for autosomal recessive polycystic kidney disease using multiple displacement amplification. J Assist Reprod Genet. 2010; 27: 397-407. [CrossRef]
- Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009; 60: 321-337. [CrossRef]
- Pei Y, Watnick T. Diagnosis and screening of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010; 17: 140-152. [CrossRef]
- Chapman AB, Devuyst O, Eckardt KU, Gansevoort RT, Harris T, Horie S, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015; 88: 17-27. [CrossRef]
- Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993; 329: 332-342. [CrossRef]
- Pretorius DH, Lee ME, Manco-Johnson ML, Weingast GR, Sedman AB, Gabow PA. Diagnosis of autosomal dominant polycystic kidney disease in utero and in the young infant. J Ultrasound Med. 1987; 6: 249-255. [CrossRef]
- MacDermot KD, Saggar-Malik AK, Economides DL, Jeffery S. Prenatal diagnosis of autosomal dominant polycystic kidney disease (PKD1) presenting in utero and prognosis for very early onset disease. J Med Genet. 1998; 35: 13-16. [CrossRef]
- Cadnapaphornchai MA. Autosomal dominant polycystic kidney disease in children. Curr Opin Pediatr. 2015; 27: 193-200. [CrossRef]
- Fick GM, Duley IT, Johnson AM, Strain JD, Manco-Johnson ML, Gabow PA. The spectrum of autosomal dominant polycystic kidney disease in children. J Am Soc Nephrol. 1994; 4: 1654-1660.
- Fick-Brosnahan G, Johnson AM, Strain JD, Gabow PA. Renal asymmetry in children with autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1999; 34: 639-645. [CrossRef]
- Brun M, Maugey-Laulom B, Eurin D, Didier F, Avni EF. Prenatal sonographic patterns in autosomal dominant polycystic kidney disease: a multicenter study. Ultrasound Obstet Gynecol. 2004; 24: 55-61. [CrossRef]
- Gabow PA. Autosomal dominant polycystic kidney disease--more than a renal disease. American Journal of Kidney Diseases. 1990; 16: 403-413. [CrossRef]
- Everson GT. Hepatic cysts in autosomal dominant polycystic kidney disease. Mayo Clinic Proceedings. 1990; 65: 1020-1025. [CrossRef]
- Bae KT, Zhu F, Chapman AB, Torres VE, Grantham JJ, Guay-Woodford LM, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the consortium for radiologic imaging studies of polycystic kidney disease cohort. Clin J Am Soc Nephrol. 2006; 1: 64-69. [CrossRef]
- Ong AC. Screening for intracranial aneurysms in ADPKD. Bmj. 2009; 339: b3763. [CrossRef]
- Cadnapaphornchai MA. Hypertension in children with autosomal dominant polycystic kidney disease (ADPKD). Curr Hypertens Rev. 2013; 9: 21-26. [CrossRef]
- Lanktree MB, Chapman AB. New treatment paradigms for ADPKD: moving towards precision medicine. Nat Rev Nephrol. 2017. [CrossRef]
- Schrier RW, Abebe KZ, Perrone RD, Torres VE, Braun WE, Steinman TI, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med. 2014. [CrossRef]
- Fick-Brosnahan GM, Tran ZV, Johnson AM, Strain JD, Gabow PA. Progression of autosomal-dominant polycystic kidney disease in children. Kidney Int. 2001; 59: 1654-1662. [CrossRef]
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med. 2008; 359: 1477-1485. [CrossRef]
- Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Prospective change in renal volume and function in children with ADPKD. Clin J Am Soc Nephrol. 2009; 4: 820-829. [CrossRef]
- Wong H, Vivian L, Weiler G, Filler G. Patients with autosomal dominant polycystic kidney disease hyperfiltrate early in their disease. Am J Kidney Dis. 2004; 43: 624-628. [CrossRef]
- Helal I, Reed B, McFann K, Yan XD, Fick-Brosnahan GM, Cadnapaphornchai M, et al. Glomerular hyperfiltration and renal progression in children with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2011; 6: 2439-2443. [CrossRef]
- Sedman A, Bell P, Manco-Johnson M, Schrier R, Warady BA, Heard ED, et al. Autosomal dominant polycystic kidney disease in childhood: a longitudinal study. Kidney Int. 1987; 31: 1000-1005. [CrossRef]
- Massella L, Mekahli D, Paripovic D, Prikhodina L, Godefroid N, Niemirska A, et al. Prevalence of hypertension in children with early-stage ADPKD. Clin J Am Soc Nephrol. 2018; 13: 874-883. [CrossRef]
- Cadnapaphornchai MA, Fick-Brosnahan GM, Duley I, Johnson AM, Strain JD, DeGroff CG, et al. Design and baseline characteristics of participants in the study of antihypertensive therapy in children and adolescents with autosomal dominant polycystic kidney disease (ADPKD). Contemp Clin Trials. 2005; 26: 211-222. [CrossRef]
- Schrier RW, Brosnahan G, Cadnapaphornchai MA, Chonchol M, Friend K, Gitomer B, et al. Predictors of autosomal dominant polycystic kidney disease progression. J Am Soc Nephrol. 2014; 25: 2399-2418. [CrossRef]
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2009; 20: 1888-1893. [CrossRef]
- Schrier RW. Hypertension and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2011; 57: 811-813. [CrossRef]
- Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Increased left ventricular mass in children with autosomal dominant polycystic kidney disease and borderline hypertension. Kidney Int. 2008; 74: 1192-1196. [CrossRef]
- Chapman AB, Guay-Woodford LM, Grantham JJ, Torres VE, Bae KT, Baumgarten DA, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int. 2003; 64: 1035-1045. [CrossRef]
- Chapman AB, Guay-Woodford LM. Renal volume in children with ADPKD: size matters. Clin J Am Soc Nephrol. 2009; 4: 698-699. [CrossRef]
- Chapman AB, Bost JE, Torres VE, Guay-Woodford L, Bae KT, Landsittel D, et al. Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2012; 7: 479-486. [CrossRef]
- Flynn JT. Ambulatory blood pressure monitoring in children: imperfect yet essential. Pediatr Nephrol. 2011; 26: 2089-2094. [CrossRef]
- Flynn JT, Falkner BE. New clinical practice guideline for the management of high blood pressure in children and adolescents. Hypertension. 2017; 70: 683-686. [CrossRef]
- Seeman T, Dusek J, Vondrichova H, Kyncl M, John U, Misselwitz J, et al. Ambulatory blood pressure correlates with renal volume and number of renal cysts in children with autosomal dominant polycystic kidney disease. Blood Press Monit. 2003; 8: 107-110. [CrossRef]
- Chapman AB, Stepniakowski K, Rahbari-Oskoui F. Hypertension in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010; 17: 153-163. [CrossRef]
- Burgmaier K, Kunzmann K, Ariceta G, Bergmann C, Buescher AK, Burgmaier M, et al. Risk factors for early dialysis dependency in autosomal recessive polycystic kidney disease. J Pediatr. 2018; 199: 22-8 e6.
- Cadnapaphornchai MA, George DM, Masoumi A, McFann K, Strain JD, Schrier RW. Effect of statin therapy on disease progression in pediatric ADPKD: design and baseline characteristics of participants. Contemp Clin Trials. 2011; 32: 437-445. [CrossRef]
- Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K, et al. Mutations in GANAB, encoding the glucosidase II alpha subunit, cause autosomal-dominant polycystic kidney and liver disease. Am J Hum Genet. 2016; 98: 1193-1207. [CrossRef]
- Cornec-Le Gall E, Olson RJ, Besse W, Heyer CM, Gainullin VG, Smith JM, et al. Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet. 2018; 102: 832-844. [CrossRef]
- Consortium TEPKD. The polycystic kidney disease1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell. 1994; 77: 881-894. [CrossRef]
- Loftus BJ, Kim UJ, Sneddon VP, Kalush F, Brandon R, Fuhrmann J, et al. Genome duplications and other features in 12 Mb of DNA sequence from human chromosome 16p and 16q. Genomics. 1999; 60: 295-308. [CrossRef]
- Hwang YH, Conklin J, Chan W, Roslin NM, Liu J, He N, et al. Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2015.
- Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995; 10: 151-160. [CrossRef]
- Gainullin VG, Hopp K, Ward CJ, Hommerding CJ, Harris PC. Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J Clin Invest. 2015; 125: 607-620. [CrossRef]
- Qian F, Boletta A, Bhunia AK, Xu H, Liu L, Ahrabi AK, et al. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1- associated mutations. Proc Natl Acad Sci U S A. 2002; 99: 16981-16986. [CrossRef]
- Kurbegovic A, Kim H, Xu H, Yu S, Cruanes J, Maser RL, et al. Novel functional complexity of polycystin-1 by GPS cleavage in vivo: role in polycystic kidney disease. Mol Cell Biol. 2014; 34: 3341-3353. [CrossRef]
- Low SH, Vasanth S, Larson CH, Mukherjee S, Sharma N, Kinter MT, et al. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev Cell. 2006; 10: 57-69. [CrossRef]
- Yu W, Kong T, Beaudry S, Tran M, Negoro H, Yanamadala V, et al. Polycystin-1 protein level determines activity of the Galpha12/JNK apoptosis pathway. J Biol Chem. 2010; 285: 10243-10251. [CrossRef]
- Lin CC, Kurashige M, Liu Y, Terabayashi T, Ishimoto Y, Wang T, et al. A cleavage product of Polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Sci Rep. 2018; 8: 2743. [CrossRef]
- Mochizuki T, Wu G, Hayashi T, Xenophontos SL, B. V, Saris JJ, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996; 272: 1339-1342. [CrossRef]
- Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, et al. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol. 2002; 4: 191-197. [CrossRef]
- Kim H, Xu H, Yao Q, Li W, Huang Q, Outeda P, et al. Ciliary membrane proteins traffic through the Golgi via a Rabep1/GGA1/Arl3-dependent mechanism. Nat Commun. 2014; 5: 5482. [CrossRef]
- Garcia-Gonzalez MA, Menezes LF, Piontek KB, Kaimori J, Huso DL, Watnick T, et al. Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum Mol Genet. 2007; 16: 1940-1950. [CrossRef]
- Happe H, Peters DJ. Translational research in ADPKD: lessons from animal models. Nat Rev Nephrol. 2014; 10: 587-601. [CrossRef]
- Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet. 2004; 13: 3069-3077. [CrossRef]
- Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG, et al. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest. 2012; 122: 4257-4273. [CrossRef]
- Tan AY, Blumenfeld J, Michaeel A, Donahue S, Bobb W, Parker T, et al. Autosomal dominant polycystic kidney disease caused by somatic and germline mosaicism. Clin Genet. 2014.
- Pei Y, Lan Z, Wang K, Garcia-Gonzalez M, He N, Dicks E, et al. A missense mutation in PKD1 attenuates the severity of renal disease. Kidney Int. 2012; 81: 412-417. [CrossRef]
- Bergmann C, von Bothmer J, Ortiz Bruchle N, Venghaus A, Frank V, Fehrenbach H, et al. Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol. 2011; 22: 2047-2056. [CrossRef]
- Brook-Carter PT, Peral B, Ward CJ, Thompson P, Hughes J, Maheshwar MM, et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease--a contiguous gene syndrome. Nat Genet. 1994; 8: 328-332. [CrossRef]
- Fedeles SV, Tian X, Gallagher AR, Mitobe M, Nishio S, Lee SH, et al. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat Genet. 2011; 43: 639-647. [CrossRef]
- Fedeles SV, Gallagher AR, Somlo S. Polycystin-1: a master regulator of intersecting cystic pathways. Trends Mol Med. 2014; 20: 251-260. [CrossRef]
- Besse W, Dong K, Choi J, Punia S, Fedeles SV, Choi M, et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest. 2017; 127: 1772-1785. [CrossRef]
- Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet. 2013; 45: 1004-1012. [CrossRef]
- Ma M, Gallagher AR, Somlo S. Ciliary Mechanisms of Cyst Formation in Polycystic Kidney Disease. Cold Spring Harb Perspect Biol. 2017; 9. [CrossRef]
- Lee SH, Somlo S. Cyst growth, polycystins, and primary cilia in autosomal dominant polycystic kidney disease. Kidney Res Clin Pract. 2014; 33: 73-78. [CrossRef]
- Wu G, D'Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998; 93: 177-188. [CrossRef]
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med. 2001; 7: 151-156. [CrossRef]
- Eccles MR, Stayner CA. Polycystic kidney disease - where gene dosage counts. F1000Prime Rep. 2014; 6: 24. [CrossRef]
- Harris PC. What is the role of somatic mutation in autosomal dominant polycystic kidney disease? J Am Soc Nephrol. 2010; 21: 1073-1076. [CrossRef]
- Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med. 2007; 13: 1490-1495. [CrossRef]
- Shibazaki S, Yu Z, Nishio S, Tian X, Thomson RB, Mitobe M, et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet. 2008. [CrossRef]
- Gallagher AR, Germino GG, Somlo S. Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010; 17: 118-130. [CrossRef]
- Rogers KA, Moreno SE, Smith LA, Husson H, Bukanov NO, Ledbetter SR, et al. Differences in the timing and magnitude of Pkd1 gene deletion determine the severity of polycystic kidney disease in an orthologous mouse model of ADPKD. Physiol Rep. 2016; 4. [CrossRef]
- Leonhard WN, Zandbergen M, Veraar K, van den Berg S, van der Weerd L, Breuning M, et al. Scattered deletion of PKD1 in kidneys causes a cystic snowball effect and recapitulates polycystic kidney disease. J Am Soc Nephrol. 2014.
- Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000; 11: 1179-1187.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004; 279: 40419-40430. [CrossRef]
- Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, et al. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int. 2004; 66: 964-973. [CrossRef]
- Gattone VH, 2nd, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. 2003; 9: 1323-1326. [CrossRef]
- Wang X, Gattone V, 2nd, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol. 2005; 16: 846-851. [CrossRef]
- Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, et al. Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease. N Engl J Med. 2012. [CrossRef]
- Torres VE, Higashihara E, Devuyst O, Chapman AB, Gansevoort RT, Grantham JJ, et al. Effect of tolvaptan in autosomal dominant polycystic kidney disease by CKD stage: Results from the TEMPO 3:4 Trial. Clin J Am Soc Nephrol. 2016; 11: 803-811. [CrossRef]
- Mustafa RA, Yu ASL. Burden of proof for tolvaptan in ADPKD: Did REPRISE provide the answer? Clin J Am Soc Nephrol. 2018; 13: 1107-1109. [CrossRef]
- Gilbert RD, Evans H, Olalekan K, Nagra A, Haq MR, Griffiths M. Tolvaptan treatment for severe neonatal autosomal-dominant polycystic kidney disease. Pediatr Nephrol. 2017; 32: 893-896. [CrossRef]
- Harskamp LR, Gansevoort RT, Boertien WE, van Oeveren W, Engels GE, van Goor H, et al. Urinary EGF receptor ligand excretion in patients with autosomal dominant polycystic kidney disease and response to tolvaptan. Clin J Am Soc Nephrol. 2015; 10: 1749-1756. [CrossRef]
- Du J, Wilson PD. Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am J Physiol. 1995; 269: C487-C495. [CrossRef]
- Pugh JL, Sweeney WE, Jr., Avner ED. Tyrosine kinase activity of the EGF receptor in murine metanephric organ culture. Kidney Int. 1995; 47: 774-781. [CrossRef]
- Orellana SA, Sweeney WE, Neff CD, Avner ED. Epidermal growth factor receptor expression is abnormal in murine polycystic kidney. Kidney Int. 1995; 47: 490-499. [CrossRef]
- Richards WG, Sweeney WE, Yoder BK, Wilkinson JE, Woychik RP, Avner ED. Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease. J Clin Invest. 1998; 101: 935-939. [CrossRef]
- Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int. 2000; 57: 33-40. [CrossRef]
- Sweeney WE, Jr, Hamahira K, Sweeney J, Garcia-Gatrell M, Frost P, Avner ED. Combination treatment of PKD utilizing dual inhibition of EGF-receptor activity and ligand bioavailability. Kidney Int. 2003; 64: 1310-1319. [CrossRef]
- Ma R, Li WP, Rundle D, Kong J, Akbarali HI, Tsiokas L. PKD2 functions as an epidermal growth factor-activated plasma membrane channel. Mol Cell Biol. 2005; 25: 8285-8298. [CrossRef]
- Aguiari G, Bizzarri F, Bonon A, Mangolini A, Magri E, Pedriali M, et al. Polycystin-1 regulates amphiregulin expression through CREB and AP1 signalling: implications in ADPKD cell proliferation. J Mol Med (Berl). 2012. [CrossRef]
- Streets AJ, Magayr TA, Huang L, Vergoz L, Rossetti S, Simms RJ, et al. Parallel microarray profiling identifies ErbB4 as a determinant of cyst growth in ADPKD and a prognostic biomarker for disease progression. Am J Physiol Renal Physiol. 2017; 312: F577-F588. [CrossRef]
- Sweeney WE, Jr., von Vigier RO, Frost P, Avner ED. Src Inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol. 2008; 19: 1331-1341. [CrossRef]
- Talbot JJ, Shillingford JM, Vasanth S, Doerr N, Mukherjee S, Kinter MT, et al. Polycystin-1 regulates STAT activity by a dual mechanism. Proc Natl Acad Sci U S A. 2011; 108: 7985-7990. [CrossRef]
- Silva CM. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene. 2004; 23: 8017-8023. [CrossRef]
- Talbot JJ, Song X, Wang X, Rinschen MM, Doerr N, Lariviere WB, et al. The Cleaved cytoplasmic tail of polycystin-1 regulates Src-dependent STAT3 activation. J Am Soc Nephrol. 2014; 25: 1737-1748. [CrossRef]
- Tesar V, Ciechanowski K, Pei Y, Barash I, Shannon M, Li R, et al. Bosutinib versus placebo for autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2017; 28: 3404-3413. [CrossRef]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006; 124: 471-484. [CrossRef]
- Edelstein CL. Mammalian target of rapamycin and caspase inhibitors in polycystic kidney disease. Clin J Am Soc Nephrol. 2008; 3: 1219-1226. [CrossRef]
- Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. 2006; 103: 5466-5471. [CrossRef]
- Zafar I, Belibi FA, He Z, Edelstein CL. Long-term rapamycin therapy in the Han:SPRD rat model of polycystic kidney disease (PKD). Nephrol Dial Transplant. 2009; 24: 2349-2353. [CrossRef]
- Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. 2010; 21: 489-497. [CrossRef]
- Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, Sommerer C, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010; 363: 830-840. [CrossRef]
- Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010; 363: 820-829. [CrossRef]
- Shillingford JM, Leamon CP, Vlahov IR, Weimbs T. Folate-conjugated rapamycin slows progression of polycystic kidney disease. J Am Soc Nephrol. 2012; 23: 1674-1681. [CrossRef]
- Sweeney WE, Frost P, Avner ED. Tesevatinib ameliorates progression of polycystic kidney disease in rodent models of autosomal recessive polycystic kidney disease. World J Nephrol. 2017; 6: 188-200. [CrossRef]
- Klawitter J, Klawitter J, McFann K, Pennington AT, Abebe KZ, Brosnahan G, et al. Bioactive lipid mediators in polycystic kidney disease. J Lipid Res. 2014; 55: 1139-1149. [CrossRef]
- Park F, Sweeney W, Jia G, Roman RJ, Avner ED. Contribution of cytochrome P450 metabolites to the abnormal proliferation of renal epithelial cells in a murine model of PKD. J Am Soc Nephrol. 2007; 18: 129A.
- Park F, Sweeney WE, Jia G, Roman RJ, Avner ED. 20-HETE mediates proliferation of renal epithelial cells in polycystic kidney disease. J Am Soc Nephrol. 2008; 19: 1929-1939. [CrossRef]
- Park F, Sweeney WE, Jr., Jia G, Akbulut T, Mueller B, Falck JR, et al. Chronic blockade of 20-HETE synthesis reduces polycystic kidney disease in an orthologous rat model of ARPKD. Am J Physiol Renal Physiol. 2009; 296: F575-F582. [CrossRef]
- Imig JD, Ryan MJ. Immune and inflammatory role in renal disease. Compr Physiol. 2013; 3: 957-976. [CrossRef]
- Wang YY, Yang YX, Zhe H, He ZX, Zhou SF. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: an update on its pharmacokinetic and pharmacodynamic properties. Drug Des Devel Ther. 2014; 8: 2075-2088.
- Patil A, Sweeney WE, Jr., Pan CG, Avner ED. Unique interstitial miRNA signature drives fibrosis in a murine model of autosomal dominant polycystic kidney disease. World J Nephrol. 2018; 7: 108-116. [CrossRef]
- Hajarnis S, Lakhia R, Patel V. MicroRNAs and Polycystic Kidney Disease. In: Li X, editor. Polycystic Kidney Disease. Brisbane (AU)2015.
- Hajarnis S, Lakhia R, Yheskel M, Williams D, Sorourian M, Liu X, et al. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun. 2017; 8: 14395. [CrossRef]
- Hajarnis S, Yheskel M, Williams D, Brefort T, Glaudemans B, Debaix H, et al. Suppression of microRNA activity in kidney collecting ducts induces partial loss of epithelial phenotype and renal fibrosis. J Am Soc Nephrol. 2017.
- Torres VE, Abebe KZ, Chapman AB, Schrier RW, Braun WE, Steinman TI, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med. 2014. [CrossRef]
- McFarlane SI, Muniyappa R, Francisco R, Sowers JR. Clinical review 145: Pleiotropic effects of statins: lipid reduction and beyond. J Clin Endocrinol Metab. 2002; 87: 1451-1458. [CrossRef]
- Wong ATY, Mannix C, Grantham JJ, Allman-Farinelli M, Badve SV, Boudville N, et al. Randomised controlled trial to determine the efficacy and safety of prescribed water intake to prevent kidney failure due to autosomal dominant polycystic kidney disease (PREVENT-ADPKD). BMJ Open. 2018; 8: e018794.
- Cadnapaphornchai MA, George DM, McFann K, Wang W, Gitomer B, Strain JD, et al. Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2014; 9: 889-896. [CrossRef]
- Warner G, Hein KZ, Nin V, Edwards M, Chini CC, Hopp K, et al. Food Restriction ameliorates the development of polycystic kidney disease. J Am Soc Nephrol. 2015.
- Murphy EL, Droher ML, DiMaio MS, Dahl NK. Preimplantation genetic diagnosis counseling in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2018; 72: 866-872. [CrossRef]
- Gendreau SB, Ventura R, Keast P, Laird AD, Yakes FM, Zhang W, et al. Inhibition of the T790M gatekeeper mutant of the epidermal growth factor receptor by EXEL-7647. Clin Cancer Res. 2007; 13: 3713-3723. [CrossRef]
- Trowe T, Boukouvala S, Calkins K, Cutler RE, Jr., Fong R, Funke R, et al. EXEL-7647 inhibits mutant forms of ErbB2 associated with lapatinib resistance and neoplastic transformation. Clin Cancer Res. 2008; 14: 2465-2475. [CrossRef]
