

Genetic and Epigenetic Regulation of Telomere Length: Current Findings, Methodological Limitations and Possibilities for Future Studies
Taylor Lawrence 1 ![]() , Therese M. Murphy 1, 2, *
, Therese M. Murphy 1, 2, * ![]()
![]()
- University of Exeter Medical School, University of Exeter, Exeter, UK
- School of Biological and Health Sciences, College of Sciences and Health, Dublin Institute of Technology, Dublin 8, Ireland
* Correspondence: Therese M. Murphy ![]()
![]()
Received: October 29, 2018 | Accepted: December 14, 2018 | Published: December 20, 2018
OBM Genetics 2018, Volume 2, Issue 4 doi: 10.21926/obm.genet.1804055
Academic Editors: Stéphane Viville and Marcel Mannens
Special Issue: Epigenetic Mechanisms in Health and Disease
Recommended citation: Lawrence T, Murphy TM. Genetic and Epigenetic Regulation of Telomere Length: Current Findings, Methodological Limitations and Possibilities for Future Studies. OBM Genetics 2018;2(4):055; doi:10.21926/obm.genet.1804055.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Telomeres are TTAGGG repeats located at the end of chromosomes that maintain DNA stability. Telomere length (TL) has been widely implicated as a marker of biological age, and is associated with several human diseases, including depression, cardiovascular disease and cancer. Twin studies and cohort studies estimate heritability of TL between 78-82%. Moreover, several genomic loci which influence TL have been identified. Despite the success of genetic studies in furthering our understanding of telomere biology, identified variants account for only a small proportion of the estimated heritability. Over the last decade, epigenetic regulation of mammalian telomeres has become apparent. These epigenetic mechanisms, which act to regulate gene expression via modifications to DNA, histone proteins and chromatin, change with age and in response to specific environmental and psychosocial factors —providing a mechanism for the interaction between genotype and the environment. The present review examines the evidence for genetic and epigenetic regulation of TL and discusses their role in human disease, before outlining some of the methodological limitations of these studies. Finally, the review defines what the ‘epigenetic clock’ is and evaluates its relationship with TL.
Keywords
Telomere; genetics; GWAS; epigenetics; DNA methylation; epigenetic clock
1. Introduction
Telomeres are ribonucleoprotein complexes that protect the ends of eukaryotic chromosomes [1] and they play a critical role in maintaining genomic stability [2]. Telomere length (TL) and sequence varies between species, but in humans the average length is 5-15kb and is made up of TTAGGG repeats [3]. The main function of telomeres is ‘capping’ the chromosomes, preventing them from damage [4]. Double stranded DNA makes up the majority of telomeres and a single-stranded DNA overhang forms the t-loop (telomeric-loop) which is supported by a network of proteins called shelterin [3]. Shelterin plays a role in protection from enzymatic digestion, TL regulation and controlling signalling cascades [5]. These properties enable telomeres to protect chromosome ends from degradation, DNA damage and fusion. DNA replication is proficient at copying coding DNA but the eukaryotic DNA replication machinery is unable to copy the end of telomeres that have a single-stranded structure [6], resulting in telomere shortening. This gradual loss of repeats reduces the renewal capacity of cells and ultimately causes the onset of cellular senescence [6]. If cells continue to divide the telomeres fuse, causing genomic instability that can lead to a number of diseases including cancer. TL has been widely implicated as a marker of biological age [7], and is influenced by inflammation and cellular stress [8]. Furthermore, shorter telomeres are robustly associated with several human diseases, including depression [9], cardiovascular disease [10] and cancer [11]. This review examines the evidence for genetic and epigenetic regulation of TL and discusses the role in human disease, before outlining some of the methodological limitations of these studies. Finally, the review defines what the ‘epigenetic clock’ is and evaluates its relationship with TL.
1.1 Telomere Shortening
TL is maintained by the action of telomerase, a ribonucleoprotein which contains the RNA template TERC and a reverse transcriptase TERT, in some cell types, notably stem and germ cells [12]. However, in most somatic tissues telomeres shorten with each cell division, a process believed to be accelerated by oxidative stress and inflammation [13,14]. In somatic cells telomeres become shortened by 100-200bp with each cell division, due to the lack of TL maintenance normally carried out by telomerase [15]. Telomerase activity in most somatic cells diminishes after birth resulting in gradual telomere shortening [16]. Since a critical TL is needed to prevent activation of DNA damage pathways, the shortening of telomeres eventually leads to cell cycle arrest, a hallmark of cellular senescence, considered to promote ageing [16].
1.2 Telomeres Roles in Human Disease
Inter-individual variation in mean TL has been associated with cancer and several age-associated diseases and TL has emerged as a promising biomarker for biological age [7]. Early evidence of the role of telomeres in disease emerged from studies of telomeropathies, which develop when telomere erosion occurs prematurely as a consequence of mutations in genes coding for factors involved in telomere maintenance and repair. Human telomeropathies include Hoyeraal-Hreidarsson syndrome (HHS), dyskeratosis congenita (DC) and aplastic anemia [17]. Although these diseases show a wide and complex range of clinical symptoms, all of them are characterized by presenting with critically short telomeres. TL is also associated with a several cancers and mutations in telomere-associated genes, such as TERT, OBFC1 and TERC, are associated with an increased risk of multiple cancers [18]. It has even been suggested that short leucocyte TL (LTL) increases the risk of all cancers and cancer fatalities [19]. Moreover, illnesses such as depression and diseases with an inflammatory etiology have been associated with telomere shortening [20,21,22]. Together with mouse models of telomerase deficiency, these observations provided important biological insight illustrating that short telomeres are a major mechanistic cause of the disease phenotypes [23]. Moreover, multiple model systems (e.g., ciliates, yeast, plants, and mouse) have been instrumental in studying the molecular underpinnings of telomere regulation and their role in disease and have been extensively reviewed elsewhere [2,24,25]. Although a causal relationship between telomerase deficiency and human disease has been established, the cause and effect relationship between variation in TL and human disease is less well understood. Deciphering the role of genetic and epigenetic regulation of TL in humans will further facilitate our understanding of telomere biology and help elucidate biological pathways that are influenced by changes in TL and advance our understanding of the biology of ageing and age-associated diseases.
1.3 Genetic Regulation of Telomere Length
Since the 1990s, it has been established that mean TL shortens with age at a rate of roughly 31bp per year [26] but variation is frequently observed between individuals of the same age, implying that something other than age can also effect TL [26]. Twin studies and cohort studies were used to investigate the mean heritability of TL which was shown to range between 78-82% [27,28]. This high heritability has led to several investigations to identify genomic loci which illicit an effect on TL.
Genome-wide association studies of telomere length. The first locus to be associated with TL was TERC, one of the components of telomerase. The first genome-wide association studies (GWAS) to identify the association between TL and genetic variation at the TERC locus was conducted in 2917 European individuals with replication in 9492 individuals, whereby a SNP (rs12696304) located 1.5kb downstream from the TERC locus was significantly associated (P=3.72 x 10-14) with shorter mean LTL [29]. Another GWAS study identified a SNP (rs10936599) within a haplotype block that encompassed the TERC locus and was significantly associated (P=3.92 x 10-5) with longer TL [30], in contrast to the previous identified TERC-associated SNP [29]. This may be due to neither SNP being located in TERCs coding region and instead they effect TERC expression through one of the other genes at the 3q26 locus, with one SNP preventing its expression and the other increasing its function [29]. Jones et al also identified that the SNP resulted in a heightened risk of developing colorectal cancer, providing evidence that TL does indeed serve a role in the development of some diseases [30]. A third GWAS study confirmed that genetic variation at the TERC locus is associated with mean LTL and was conducted in 3,417 participants with replication through de-novo genotyping [31]. They also identified a SNP (rs4387287) in the OBFC1 (STN1) gene, that is significantly associated (P=3.9 x 10-9) with mean LTL [31]. The protein encoded by OBFC1 stimulates DNA polymerase-alpha-primase, the enzyme that initiates DNA replication [32] and appears to function in a telomere-associated complex with C17ORF68 and TEN1 [33].
TERT or telomerase reverse transcriptase codes for the catalytic subunit of telomerase and forms the telomerase enzyme with TERC [34] (See Figure 1). A study identified two independent SNPs (rs2736108 P=5.8 x 10-7. rs7705526 P=2.3 x 10-14), located in the TERT promoter and TERT intron 2 respectively, that are both significantly associated with longer TL [35]. The SNP (rs7705526) located in TERT intron 2 was also significantly associated (P=1.3 x 10-15) with a higher risk of low malignant potential ovarian cancer [35]. Multiple different studies have attempted to replicate this finding, and whilst some have been successful Shen et al found no genome-wide significant associations between TERT and TL [36]. It is possible that this is due to the small sample size of the study (n=742). Despite the power of these approaches to identify genetic variation associated with TL, these initial studies were moderately powered to identify genetic variants associated with TL and more recent genetic studies have employed large-scale meta-analyses to identify robust genetic loci associated with TL.
Meta-analyses investigating genetic loci associated with TL. The first large-scale TL GWAS meta-analysis to be conducted included 9190 participants from six GWASs (European Population) with replication in 2226 participants from four studies [37]. This study confirmed the association between TL and genetic variation at the previously identified OBFC1 and TERC loci (OBFC1, rs9419958 P=9.1 × 10−11. TERC, rs1317082 P=1.1 × 10−8) [37]. The meta-analysis also identified novel genetic loci associated with TL, CTC1 (rs3027234 P=3.6 x 10-8) and ZNF676 (rs412658 P=3.3 x 10-8) [37]. The OBFC1 and CTC1 loci together make up 2/3 of the human heterotrimer complex named CST (for Cdc13-Stn1-Ten1) suggesting that it may play an important role in maintaining telomeres [37]. In 2013, the largest GWAS meta-analysis of LTL was performed with a cohort of 37,684 individuals and replication in 10,739 individuals. The study identified 7 robust genetic loci-that were significantly associated with mean LTL (P < 5 x 10-8) [18]. The previously identified TERC, TERT, OBFC1 loci and the novel NAF1 and RTEL1 loci [18], have a known role in the maintenances of telomeres (See Figure 1). NAF1 encodes a protein required for H/ACA box snoRNA assembly and therefore is required for the assembly of TERC as it belongs to that RNA family [38]. RTEL1 encodes a DNA helicase with roles in telomere maintenance and DNA repair [39]. In addition, the large-scale meta-analysis identified LTL-associated genetic variants associated with ZNF208 - a gene that codes for a member of the Zinc finger protein family and thus plays an important role in DNA binding mediated gene regulation (see Figure 1). The final LTL-associated genetic variant (rs11125529) is associated with both ACYP2 and TSPYL6 genes. ACYP2 has also been reported to play an important role in pyruvate metabolism [40]. The biological function of TSPYL6 is not known, but TSPYL2, another member of TSPY-like/SET/nucleosome assembly protein-1 superfamily, plays a role in chromatin remodelling and suppression of tumour growth [40]. Interestingly, the authors show that alleles associated with shorter TL are also associated with an increased risk of coronary heart disease, again supporting the idea that TL plays a role in disease [18]. More recent larger GWAS studies of TL have further built on the findings by Codd and colleagues, and successfully identified two additional robust genetic variants (DCAF4 [41] and PXK [42]) associated with TL. DCAF4 is shown to interact with DDB1 and CUL4 and is consequently suggested to possibly play a role in UVR- induced DNA damage and transcription coupled repair pathways [41]. PXK is a serine/ threonine kinase that regulates electrical excitability and it therefore seems unlikely that it would be involved in the regulation of TL. A comprehensive list of genetic variants associated with LTL is summarised in Table 1 and illustrated in Figure 1.
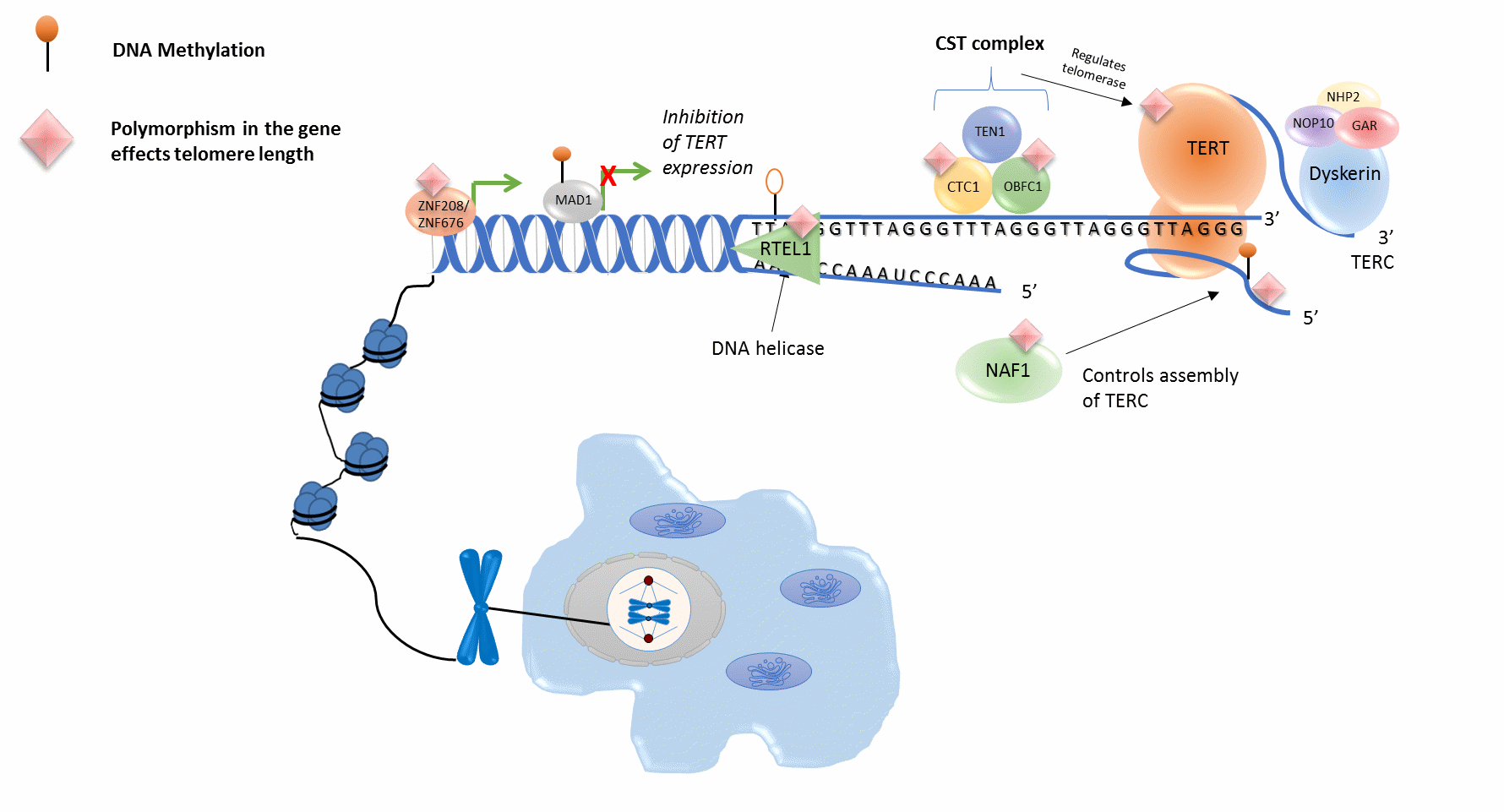
Figure 1 Molecular pathways involved in telomere Length. Several genes that are known to code for enzymes that play an important role in the regulation of telomere length exhibit genetic (e.g. CTC1, OBFC1, TERT, TERC, ZNF208, ZNF676) and epigenetic (e.g. MAD1) alterations. CST complex: cellular multiprotein complex.
Table 1 Genes associated with telomere length.

Details of the genes function, loci, associated ethnicity, SNPs and health conditions related to genetic changes in the gene are included. Genes identified in studies with less than 500 participants and no replication were excluded. All information in the table is from gene ontology, genetics home reference and NCBI websites.
Interestingly, genetic risk score analysis showed that inheritance of multiple alleles associated with shorter LTL is associated with an increased risk of coronary artery disease, providing preliminary evidence that telomere shortening might play a causal role in this condition [18].
Genome-wide associations in other ethnic groups. Many studies examining genetic variation associated with TL have been conducted in European/Caucasian cohorts, and more recent studies have attempted to replicate these findings in different populations. The TERC locus has also been shown to be associated with TL in a Chinese Han [43] population and the TERT locus has been shown to be associated with TL in both Chinese Han [44] and Korean populations [45]. Several novel TL-associated genetic variants have been identified in non-european populations (see Table 1 for more details) highlighting the importance of conducting such studies in different populations to identify novel genetic loci and provide new insights into the mechanisms involved in maintaining telomeres. This is evident in a meta-analysis conducted in a Punjabi Sikh cohort that identified an intronic variant (rs74019828) in CSNK2A2, a novel gene, was associated with TL (P=4.5 x 10-8) [46]. To date, few studies have been conducted in African populations and those that have been conducted are simply comparisons to European studies [47]. In order to better understand the mechanisms by which TL is controlled future studies conducted in African populations would be beneficial, particularly as research suggests that African Americans and sub-Saharan Africans have longer LTL than Europeans and the cause of this difference is unknown [47,48] .
Parent-to-offspring transmission of telomeres. As discussed above TL is a heritable trait with two potential sources of heritability: inherited genetic variation (e.g. SNPs affecting telomere maintenance (see Table 1)) and variability in the lengths of telomeres in gametes that produce offspring zygotes [49]. The latter, is described as “direct” transmission of telomeres [50]. Evidence supports a role for parental TL on offspring TL and suggests that sperm TL increases with age in humans, and as a result offspring of older fathers inherit longer telomeres [51]. However, telomeres undergo a “reprogramming” event during early embryogenesis and it remains unclear to what extent reprogramming alters the impact of germ cell TL on offspring TL [50]. Recent observations provide evidence that TL in parental germ cells impacts TL in offspring cells and contributes to LTL heritability despite telomere “reprogramming” during embryogenesis [49]. However, larger studies are required to provide a robust estimation of LTL heritability by ‘direct’ transmission.
1.4 Epigenetic Regulation of Telomere Length
Despite the success of genetic studies in furthering our understanding of telomere biology, identified variants only account for only a small proportion of the estimated heritability. Over the last decade, epigenetic regulation of mammalian telomeres has become apparent [52,53,54]. Epigenetics can be defined as the mechanisms that initiate and maintain heritable patterns of gene expression without altering the sequence of the genome. There are several layers of epigenetic complexity including histone modifications, chromatin remodelling, micro-RNAs and DNA methylation. The latter being the most thoroughly studied to date [55]. The epigenome is potentially malleable – changing with age and in response to a plethora of environmental and psychosocial factors [56], thus providing a mechanism mediating the interaction between genetic susceptibility and environmental risk exposures [57]. Given that environmental factors (such as smoking, stress and obesity) have been shown to both influence a person’s epigenome [58,59,60] and accelerate the rate of telomere shortening [61,62] it is plausible that together these changes could influence the expression of subtelomeric genes.
The most widely studied epigenetic modification is DNA methylation, which refers to the covalent addition of a methyl group to the carbon at position 5 of the cytosine ring, by a family of DNA methyltransferase (DNMT) enzymes, resulting in 5-methylcytosine [63]. In mammalian DNA, 5-methylcytosine is found in ∼4% of genomic DNA, primarily at cytosine–guanine dinucleotides [64]. These CpG sites are non-randomly dispersed throughout the genome, concentrated in hot spots or CpG-rich regions known as CpG islands. Approximately half of all human genes are estimated to contain a 5′ or promoter CpG island [65,66]. In contrast, mammalian telomeres consist solely of TTAGGG repeats and they lack CpG sites [2]. However, the subtelomeric region (segment of DNA adjacent to telomeric repeats) has been shown to be heavily methylated in both mice and human studies [67,68].The use of knockout mice models found that DNA methyltransferases (DNMTs) [67] and other epigenetic enzymes (e.g. histone methyltransferases (HMTs)[54]) were essential for TL regulation , highlighting a crucial role for epigenetic mechanisms in TL homeostasis in mammals.
Global and genome-wide DNA methylation studies and their association with telomere length. A study by Wong et al [69], examined the association between global DNA methylation and TL in a longitudinal study. As a proxy for global DNA methylation the study examined DNA methylation at LINE-1 and Alu elements using bisulfite pyrosequencing – a method shown to accurately reflect global DNA methylation levels [70]. The study found an association between both LINE-1 (P < 0.01) and Alu methylation (P = 0.02) and TL, after controlling for confounders such as age, smoking and cellular heterogeneity. In addition, the rate of telomeric change was shown to be correlated with the quantity of LINE-1 methylation [69]. Although the sample size was limited, this exploratory study suggests global DNA hypomethylation may be related to decreased TL in peripheral blood leukocytes. A recent larger study using an ELISA-based method to determine global 5mC content also showed that decreased global DNA methylation was associated with shortened TL in adolescents [71]. Taken together these studies provide the impetus for future studies, involving larger cohorts, to focus on gene-specific DNA methylation signatures potentially related to TL. Such studies are likely to further our understanding of the role telomeres play in the etiology of chronic diseases [69].
Buxton et al [12], performed the first genome-wide DNA methylation association study of TL in a small cohort (n = 24), using the Illumina 450k Beadchip (450k) array. The study identified 65 gene promoters that were enriched for CpG sites where methylation levels were associated with TL [12]. Interestingly, TL-associated epigenetics changes were enriched for subtelomeric and imprinted genomic regions [12]. Several of the 65 loci enriched for CpG sites at which DNA methylation levels are associated with TL have potential roles in human telomere biology, such as the MAD1L1 gene, a potent regulator of TERT (See Figure 1) [12]. These results point towards a bi-directional relationship between epigenetics and telomere shortening. Whereby as telomeres shorten, the methylation levels of many subtelomeric-associated genes change. This potentially results in transcriptional changes to these genes and therefore DNA methylation changes at telomere-associated genes, which may further contribute to dysregulation of TL in humans. The latter is further supported by studies in cancer, which have shown that the epigenetic plasticity of the TERT gene promoter is an important regulator of telomerase activity [72,73]. Moreover, inhibiting the expression of the TERT gene through epigenetic mechanisms can lead to telomeric shortening. Both DNA hypermethylation and histone modifications of the hTERT promoter have been associated with TERT expression and TL shortening [73]. A major limitation of this study is the small sample size used. In 2016, Marioni and colleagues, performed a large-scale meta-analysis of epigenome-wide association studies (EWAS) of TL [74] in old-age cohorts (n = 2194). Marioni and colleagues did not identify any robust DNA methylation changes associated with TL. Of note, the cohorts used in the Marioni study were from participants > 70 years of age and it would be interesting to repeat such analysis in younger cohorts, whose epigenome and telomere biology will be less influenced by age-related diseases.
Telomere position effect. Telomere shortening can also alter the expression of nearby genes, a phenomenon known as the telomere position effect (TPE) [75]. TPE involves the spreading of telomeric heterochromatin resulting in transcriptional silencing of nearby genes [76]. To date, most studies of TPE are limited to model organisms (e.g Yeast), where telomeres have been shown to loop over longer distances and repress genes up to 20 kb from the telomere end [77,78]. In human studies, chromosome looping has been shown to enable telomeres to access distant genetic loci (up to 10 mb away) in their respective chromosomes and affect their silencing. Interestingly, the same loci become separated when telomeres are shortened [76]. This process is called telomere position effect over long distances (TPE-OLD) and research suggests that this phenomenon produces extensive changes in gene regulation before telomere shortening induces DNA damage signals [76]. Further investigation of epigenetic mechanisms (e.g. chromatin modifications and DNA modifications) involved in TPE-OLD is vital to further our understanding of this phenomena and its role in diseases of ageing.
Telomere regulation by non-coding RNAs. TERRA (telomere repeat-containing RNA) is an long non-coding RNA lncRNA transcribed from the telomeric sequence [79] and have been hypothesized to regulate telomere replication and is possibly involved in silencing effects observed in TPE-OLD. The transcription of TERRA molecules is thought to be driven by DNA methylation of CpG island promoters found on subtelomeres [80]. Once transcribed TERRA molecules bind to telomerase and regulates its activity [81]. This ‘TERRA-silencing’ is suspected as a potential mechanism of telomere maintenance in human tumours through the promotion-of Alternative Lengthening of Telomeres (ALT) rather than extension by telomerase [82]. However, TERRA molecules are also implicated in cancer cells that are negative to telomerase indicating they may also act via an alternative mechanism, possibly via a DNA-damage-response (DDR) pathway [83]. Further exploration of TERRA as an epigenetic method of controlling TL is needed due to our current limited understanding of their mechanisms.
1.5 The Epigenetic Clock and Its Association with TL
Another hypothesized proxy of biological age is the epigenetic clock (or DNA methylation age (DNAm age)) [84]. The epigenetic clock measures the cumulative effect of an epigenetic maintenance system [84] by measuring DNA methylation levels at 353 CpG sites and is highly correlated with chronological age [84]. The difference between the predicted DNA methylation age and the chronological age is termed “Epigenetic age acceleration” [85]. This acceleration is correlated with several ageing related phenotypes including frailty, cancer, cardiovascular disease and Parkinson’s disease [85,86,87]. Interestingly, TL and the epigenetic clock are independently associated with chronological age [88,89]. A recent meta-analysis showed that among several estimates of epigenetic age acceleration, one measure, i.e., extrinsic epigenetic age acceleration (EEAA), was superior in predicting all-cause mortality [90]. EEAA is defined as the weighted average of DNAm age and imputed proportions of naïve CD8+ T cells, memory CD8+ T cells and plasmablasts [91]. Interestingly, LTL and EEAA are negatively correlated and that this correlation reflects the ageing of the immune system, namely the age-dependent change of the proportions of naive CD8+ T cells and memory CD8+ T cells [92]. This study suggests that DNAm Age is a proxy for immune ageing and that telomere shortening partially reflects this immune ageing. The authors suggest that as the immune system is largely developed during early life [93], studies should focus on LTL and epigenetic age in children to gain further mechanistic insights [92]. In the future, it may be interesting to combine both the epigenetic age and TL as a biomarker panel for biological ageing, and prediction of diseases of ageing.
1.6 Genetic and Epigenetic Regulation of Telomere Length: Study Design Issues
Technological caveats. Three common techniques employed in the genetic and epigenetic variation studies described above are real-time quantitative PCR (qPCR), southern blotting (SB) and single TL analysis (STELA). qPCR is the most commonly adopted method due to its ease of use and high throughput capabilities. It does have limitations as it is unable to directly measure TL in base pairs, instead, TL is measured as the ratio of telomeric product/single copy gene product (T/S) [94]. However, the changes seen in TL are normally small, meaning that precision in TL measurements may be important to obtain reproducible results [95]. Hence, SB and STELA are used due to their high precision [96]. Adoption of STELA is limited as the assays have only been developed for a small number of chromosome ends due to the lack of current knowledge on subtelomeric regions [97]. A study was consequently conducted to investigate the reproducibility of these three techniques and concluded that the variation in results between techniques was minimal with qPCR and SB having very similar reproducibility [98]. This indicates that the choice of technique has little effect on results; however, it is also important to consider how the techniques vary in methodology. For example, qPCR requires a smaller amount of DNA than the alternative techniques, allowing more samples to be compared, making it the better choice for large epidemiological studies [95].
Controlling for cellular heterogeneity. Both the epigenome and TL are confounded by cellular heterogeneity and current studies assessing average TL are often performed in bulk cells and do not account for the heterogeneity of TL among individual cells [99]. A major concern in epigenetic epidemiology or telomere studies is that any apparent disease-associated differences may simply reflect differences in cellular composition [100]. For whole blood, routine cell counts or the use of algorithms that can infer cellular composition from epigenomic data [101] can be applied to control for this variation statistically in studies examining DNA methylation changes associated with TL. Recently, a simple and robust method of Single Cell Amplification of Telomere Repeats by PCR (SCATR-PCR) has been developed [99] that can compare relative TL in individual cells based on a real-time PCR technique. SCATR-PCR coupled with advances in single cell epigenomic technology will allow future studies to directly examine epigenetic changes and TL at the single cell level.
Causality. Currently it remains unclear whether the epigenetic changes of telomere-associated genes cause or are a consequence of telomere shortening. None of the studies outlined in this review have attempted to examine the causal pathways between epigenetics changes and TL, either statistically or experimentally. Longitudinal studies (measuring both epigenetic variation and TL at multiple time points throughout the life course) and Mendelian randomization (MR) approaches are required to fully understand the temporal sequence of events. Longitudinal studies are expensive and thus MR approaches are increasingly proving to be a promising approach. MR theorises that if a trait is causally related to a phenotype, genetic variant(s) controlling activity of that trait (e.g. DNA methylation) should also be associated with the outcome (TL) [102]. Using genetic variants as proxy for exposure overcomes confounding because genetic variants are inherited at random during meiosis, so they are unrelated to potential confounders (measured or unmeasured). Therefore, following the principles of MR may represent a valid method for revealing the role of environmentally induced epigenetic changes as modifiers/risk factors of telomere shortening. Future studies examining epigenetic alterations and TL should be undertaken by applying these novel approaches to investigate the direction of effect. For instance, the 2-step epigenetic MR approach, which is an extension of MR assumptions, allows researchers to investigate the causal role of DNA methylation in the association between an environmental exposure and TL.
Additional considerations. As mentioned previously TL decreases at roughly 31bp per year [26] and TL is longer in women than in men [103]. It is consequently important when investigating TL that the findings are adjusted for both age and sex which is done in most of the studies reported above. Some also adjusted for confounding factors specific to their study including BMI and smoking, further increasing the power of these studies.
1.7 Future Perspectives
A multi-omics approach is required to disentangle the molecular basis underlying telomere biology and its association with disease. Genome-wide investigations have demonstrated that the genetic predisposition to TL and its association with disease is highly polygenic. By integrating polygenic risk scores for TL and DNA methylation profiling together would allow the research community to gain a broader and deeper understanding of the pathways primarily involved in telomere biology and their potential co-ordinated interaction. Future studies should also examine the transcriptional consequences of the observed telomere-associated DNA methylation changes. System biology methods, such as weighted correlation network analysis (WGCNA), could be applied to identify telomere-associated gene pathways and networks that could be further targeted as biomarkers or therapeutic targets for diseases of ageing.
Acknowledgement
Therese M. Murphy would like to acknowledge funding from the Brain and Behaviour research foundation through a NARSAD Young Investigator Award.
Author Contributions
Therese M. Murphy conceived the topic of the review. Taylor Lawrence prepared the first draft of the review and Therese M. Murphy edited subsequent versions of the draft.
Conflict of Interest
The authors have declared that no competing interests exist.
References
- Organization WH. Depression 2017. Available from: http://www.who.int/mediacentre/factsheets/fs369/en/.
- Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med. 2006; 12: 1133-1138. [CrossRef]
- Stewart JA, Chaiken MF, Wang F, Price CM. Maintaining the end: roles of telomere proteins in end-protection, telomere replication and length regulation. Mutat Res. 2012; 730: 12-19. [CrossRef]
- Longhese MP. DNA damage response at functional and dysfunctional telomeres. Genes Dev. 2008; 22: 125-140. [CrossRef]
- O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 2010; 11: 171-181. [CrossRef]
- Olovnikov AM. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol. 1973; 41: 181-190. [CrossRef]
- Aubert G, Lansdorp PM. Telomeres and aging. Physiol Rev. 2008; 88: 557-579. [CrossRef]
- Ridout SJ, Ridout KK, Kao HT, Carpenter LL, Philip NS, Tyrka AR, et al. Telomeres, early-life stress and mental illness. Adv Psychosom Med. 2015; 34: 92-108. [CrossRef]
- Schutte NS, Malouff JM. The association between depression and leukocyte telomere length: a meta-analysis. Depress Anxiety. 2015; 32: 229-238. [CrossRef]
- Raschenberger J, Kollerits B, Titze S, Kottgen A, Barthlein B, Ekici AB, et al. Association of relative telomere length with cardiovascular disease in a large chronic kidney disease cohort: the GCKD study. Atherosclerosis. 2015; 242: 529-534. [CrossRef]
- Ma H, Zhou Z, Wei S, Liu Z, Pooley KA, Dunning AM, et al. Shortened telomere length is associated with increased risk of cancer: a meta-analysis. PLoS One. 2011; 6: e20466. [CrossRef]
- Buxton JL, Suderman M, Pappas JJ, Borghol N, McArdle W, Blakemore AI, et al. Human leukocyte telomere length is associated with DNA methylation levels in multiple subtelomeric and imprinted loci. Sci Rep. 2014; 4: 4954. [CrossRef]
- von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002; 27: 339-344. [CrossRef]
- Kurz DJ, Decary S, Hong Y, Trivier E, Akhmedov A, Erusalimsky JD. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J Cell Sci. 2004; 117: 2417-2426. [CrossRef]
- O’Sullivan JN, Bronner MP, Brentnall TA, Finley JC, Shen WT, Emerson S, et al. Chromosomal instability in ulcerative colitis is related to telomere shortening. Nat Genet. 2002; 32: 280-284. [CrossRef]
- Hiyama E, Hiyama K. Telomere and telomerase in stem cells. Br J Cancer. 2007; 96: 1020-1024. [CrossRef]
- Martinez P, Blasco MA. Telomere-driven diseases and telomere-targeting therapies. J Cell Biol. 2017; 216: 875-887. [CrossRef]
- Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013; 45: 422-427. [CrossRef]
- Willeit P, Willeit J, Mayr A, Weger S, Oberhollenzer F, Brandstatter A, et al. Telomere length and risk of incident cancer and cancer mortality. JAMA. 2010; 304: 69-75. [CrossRef]
- Amsellem V, Gary-Bobo G, Marcos E, Maitre B, Chaar V, Validire P, et al. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011; 184: 1358-1366. [CrossRef]
- Henje Blom E, Han LK, Connolly CG, Ho TC, Lin J, LeWinn KZ, et al. Peripheral telomere length and hippocampal volume in adolescents with major depressive disorder. Transl Psychiatry. 2015; 5: e676. [CrossRef]
- Mamdani F, Rollins B, Morgan L, Myers RM, Barchas JD, Schatzberg AF, et al. Variable telomere length across post-mortem human brain regions and specific reduction in the hippocampus of major depressive disorder. Transl Psychiatry. 2015; 5: e636. [CrossRef]
- Blackburn EH, Epel ES, Lin J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science. 2015; 350: 1193-1198. [CrossRef]
- Xie Z, Jay KA, Smith DL, Zhang Y, Liu Z, Zheng J, et al. Early telomerase inactivation accelerates aging independently of telomere length. Cell. 2015; 160: 928-939. [CrossRef]
- Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012; 13: 693-704. [CrossRef]
- Hastie ND, Dempster M, Dunlop MG, Thompson AM, Green DK, Allshire RC. Telomere reduction in human colorectal carcinoma and with ageing. Nature. 1990; 346: 866-868. [CrossRef]
- Slagboom PE, Droog S, Boomsma DI. Genetic determination of telomere size in humans: a twin study of three age groups. Am J Hum Genet. 1994; 55: 876-882.
- Vasa-Nicotera M, Brouilette S, Mangino M, Thompson JR, Braund P, Clemitson JR, et al. Mapping of a major locus that determines telomere length in humans. Am J Hum Genet. 2005; 76: 147-151. [CrossRef]
- Codd V, Mangino M, van der Harst P, Braund PS, Kaiser M, Beveridge AJ, et al. Common variants near TERC are associated with mean telomere length. Nat Genet. 2010; 42: 197-199. [CrossRef]
- Jones AM, Beggs AD, Carvajal-Carmona L, Farrington S, Tenesa A, Walker M, et al. TERC polymorphisms are associated both with susceptibility to colorectal cancer and with longer telomeres. Gut. 2012; 61: 248-254. [CrossRef]
- Levy D, Neuhausen SL, Hunt SC, Kimura M, Hwang SJ, Chen W, et al. Genome-wide association identifies OBFC1 as a locus involved in human leukocyte telomere biology. Proc Natl Acad Sci U S A. 2010; 107: 9293-9298. [CrossRef]
- Casteel DE, Zhuang S, Zeng Y, Perrino FW, Boss GR, Goulian M, et al. A DNA polymerase-{alpha}{middle dot}primase cofactor with homology to replication protein A-32 regulates DNA replication in mammalian cells. J Biol Chem. 2009; 284: 5807-5818. [CrossRef]
- Miyake Y, Nakamura M, Nabetani A, Shimamura S, Tamura M, Yonehara S, et al. RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell. 2009; 36: 193-206. [CrossRef]
- Chan SW, Blackburn EH. Telomerase and ATM/Tel1p protect telomeres from nonhomologous end joining. Mol Cell. 2003; 11: 1379-1387. [CrossRef]
- Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013; 45: 371-384. [CrossRef]
- Shen J, Terry MB, Liao Y, Gurvich I, Wang Q, Senie RT, et al. Genetic variation in telomere maintenance genes, telomere length and breast cancer risk. PLoS One. 2012; 7: e44308. [CrossRef]
- Mangino M, Hwang SJ, Spector TD, Hunt SC, Kimura M, Fitzpatrick AL, et al. Genome-wide meta-analysis points to CTC1 and ZNF676 as genes regulating telomere homeostasis in humans. Hum Mol Genet. 2012; 21: 5385-5394. [CrossRef]
- Egan ED, Collins K. An enhanced H/ACA RNP assembly mechanism for human telomerase RNA. Mol Cell Biol. 2012; 32: 2428-2439. [CrossRef]
- Vannier JB, Sarek G, Boulton SJ. RTEL1: functions of a disease-associated helicase. Trends Cell Biol. 2014; 24: 416-425. [CrossRef]
- Liang Y, Zhang R, Zhang S, Ji G, Shi P, Yang T, et al. Association of ACYP2 and TSPYL6 genetic polymorphisms with risk of ischemic stroke in Han Chinese population. Mol Neurobiol. 2017; 54: 5988-5995. [CrossRef]
- Mangino M, Christiansen L, Stone R, Hunt SC, Horvath K, Eisenberg DT, et al. DCAF4, a novel gene associated with leucocyte telomere length. J Med Genet. 2015; 52: 157-162. [CrossRef]
- Pooley KA, Bojesen SE, Weischer M, Nielsen SF, Thompson D, Amin Al Olama A, et al. A genome-wide association scan (GWAS) for mean telomere length within the COGS project: identified loci show little association with hormone-related cancer risk. Hum Mol Genet. 2013; 22: 5056-5064. [CrossRef]
- Shen Q, Zhang Z, Yu L, Cao L, Zhou D, Kan M, et al. Common variants near TERC are associated with leukocyte telomere length in the Chinese Han population. Eur J Hum Genet. 2011; 19: 721-723. [CrossRef]
- Liu Y, Cao L, Li Z, Zhou D, Liu W, Shen Q, et al. A genome-wide association study identifies a locus on TERT for mean telomere length in Han Chinese. PLoS One. 2014; 9: e85043. [CrossRef]
- Do SK, Yoo SS, Choi YY, Choi JE, Jeon HS, Lee WK, et al. Replication of the results of genome-wide and candidate gene association studies on telomere length in a Korean population. Korean J Intern Med. 2015; 30: 719-726. [CrossRef]
- Saxena R, Bjonnes A, Prescott J, Dib P, Natt P, Lane J, et al. Genome-wide association study identifies variants in casein kinase II (CSNK2A2) to be associated with leukocyte telomere length in a Punjabi Sikh diabetic cohort. Circ Cardiovasc Genet. 2014; 7: 287-295. [CrossRef]
- Hansen ME, Hunt SC, Stone RC, Horvath K, Herbig U, Ranciaro A, et al. Shorter telomere length in Europeans than in Africans due to polygenetic adaptation. Hum Mol Genet. 2016; 25: 2324-2330. [CrossRef]
- Hunt SC, Chen W, Gardner JP, Kimura M, Srinivasan SR, Eckfeldt JH, et al. Leukocyte telomeres are longer in African Americans than in whites: the National Heart, Lung, and Blood Institute Family Heart Study and the Bogalusa Heart Study. Aging Cell. 2008; 7: 451-458. [CrossRef]
- Delgado DA, Zhang C, Demanelis K, Chen LS, Gao J, Roy S, et al. The contribution of parent-to-offspring transmission of telomeres to the heritability of telomere length in humans. bioRxiv. 2018. [CrossRef]
- De Meyer T, Vandepitte K, Denil S, De Buyzere ML, Rietzschel ER, Bekaert S. A non-genetic, epigenetic-like mechanism of telomere length inheritance? Eur J Hum Genet. 2014; 22: 10-11. [CrossRef]
- Broer L, Codd V, Nyholt DR, Deelen J, Mangino M, Willemsen G, et al. Meta-analysis of telomere length in 19,713 subjects reveals high heritability, stronger maternal inheritance and a paternal age effect. Eur J Hum Genet. 2013; 21: 1163-1168. [CrossRef]
- Blasco MA. The epigenetic regulation of mammalian telomeres. Nat Rev Genet. 2007; 8: 299-309. [CrossRef]
- Love WK, Berletch JB, Andrews LG, Tollefsbol TO. Epigenetic regulation of telomerase in retinoid-induced differentiation of human leukemia cells. Int J Oncol. 2008; 32: 625-631. [CrossRef]
- Garcia-Cao M, O'Sullivan R, Peters AH, Jenuwein T, Blasco MA. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat Genet. 2004; 36: 94-99. [CrossRef]
- Esteller M. The necessity of a human epigenome project. Carcinogenesis. 2006; 27: 1121-1125. [CrossRef]
- Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012; 13: 97-109. [CrossRef]
- Murphy TM, Crawford B, Dempster EL, Hannon E, Burrage J, Turecki G, et al. Methylomic profiling of cortex samples from completed suicide cases implicates a role for PSORS1C3 in major depression and suicide. Transl Psychiatry. 2017; 7: e989. [CrossRef]
- Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aissi D, Wahl S, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet. 2014; 383: 1990-1998. [CrossRef]
- Zannas AS, Provencal N, Binder EB. Epigenetics of posttraumatic stress disorder: current evidence, challenges, and future directions. Biol Psychiatry. 2015; 78: 327-335. [CrossRef]
- Li S, Wong EM, Bui M, Nguyen TL, Joo JE, Stone J, et al. Causal effect of smoking on DNA methylation in peripheral blood: a twin and family study. Clin Epigenetics. 2018; 10: 18. [CrossRef]
- Stein JY, Levin Y, Uziel O, Abumock H, Solomon Z. Traumatic stress and cellular senescence: The role of war-captivity and homecoming stressors in later life telomere length. J Affect Disord. 2018; 238: 129-135. [CrossRef]
- Mather KA, Jorm AF, Parslow RA, Christensen H. Is telomere length a biomarker of aging? A review. J Gerontol A Biol Sci Med Sci. 2011; 66: 202-213. [CrossRef]
- Razin A, Riggs AD. DNA methylation and gene function. Science. 1980; 210(4470): 604-610. [CrossRef]
- Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005; 2 Suppl 1: S4-11. [CrossRef]
- Larsen F, Gundersen G, Lopez R, Prydz H. CpG islands as gene markers in the human genome. Genomics. 1992; 13: 1095-1107. [CrossRef]
- Wang Y, Leung FC. An evaluation of new criteria for CpG islands in the human genome as gene markers. Bioinformatics. 2004; 20: 1170-1177. [CrossRef]
- Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M, et al. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol. 2006; 8: 416-424. [CrossRef]
- Vera E, Canela A, Fraga MF, Esteller M, Blasco MA. Epigenetic regulation of telomeres in human cancer. Oncogene. 2008; 27: 6817-6833. [CrossRef]
- Wong JY, De Vivo I, Lin X, Grashow R, Cavallari J, Christiani DC. The association between global DNA methylation and telomere length in a longitudinal study of boilermakers. Genet Epidemiol. 2014; 38: 254-264. [CrossRef]
- Kurdyukov S, Bullock M. DNA methylation analysis: choosing the right method. Biology (Basel). 2016; 5. [CrossRef]
- Dong Y, Huang Y, Gutin B, Raed A, Dong Y, Zhu H. Associations between Global DNA Methylation and Telomere Length in Healthy Adolescents. Sci Rep. 2017; 7: 4210. [CrossRef]
- Zinn RL, Pruitt K, Eguchi S, Baylin SB, Herman JG. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res. 2007; 67: 194-201. [CrossRef]
- Sui X, Kong N, Wang Z, Pan H. Epigenetic regulation of the human telomerase reverse transciptase gene: A potential therapeutic target for the treatment of leukemia (Review). Oncol Lett. 2013; 6: 317-322. [CrossRef]
- Marioni RE, Harris SE, Shah S, McRae AF, von Zglinicki T, Martin-Ruiz C, et al. The epigenetic clock and telomere length are independently associated with chronological age and mortality. Int J Epidemiol. 2018; 47: 356. [CrossRef]
- Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell. 1990; 63: 751-762. [CrossRef]
- Robin JD, Ludlow AT, Batten K, Magdinier F, Stadler G, Wagner KR, et al. Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances. Genes Dev. 2014; 28: 2464-2476. [CrossRef]
- Stavenhagen JB, Zakian VA. Yeast telomeres exert a position effect on recombination between internal tracts of yeast telomeric DNA. Genes Dev. 1998; 12: 3044-3058. [CrossRef]
- Tham WH, Zakian VA. Transcriptional silencing at Saccharomyces telomeres: implications for other organisms. Oncogene. 2002; 21: 512-521. [CrossRef]
- Schoeftner S, Blasco MA. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat Cell Biol. 2008; 10: 228-236. [CrossRef]
- Nergadze SG, Farnung BO, Wischnewski H, Khoriauli L, Vitelli V, Chawla R, et al. CpG-island promoters drive transcription of human telomeres. RNA. 2009; 15: 2186-2194. [CrossRef]
- Redon S, Reichenbach P, Lingner J. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res. 2010; 38: 5797-5806. [CrossRef]
- Ng LJ, Cropley JE, Pickett HA, Reddel RR, Suter CM. Telomerase activity is associated with an increase in DNA methylation at the proximal subtelomere and a reduction in telomeric transcription. Nucleic Acids Res. 2009; 37: 1152-1159. [CrossRef]
- Cusanelli E, Chartrand P. Telomeric repeat-containing RNA TERRA: a noncoding RNA connecting telomere biology to genome integrity. Front Genet. 2015; 6: 143. [CrossRef]
- Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14: R115. [CrossRef]
- Breitling LP, Saum KU, Perna L, Schottker B, Holleczek B, Brenner H. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin Epigenetics. 2016; 8: 21. [CrossRef]
- Perna L, Zhang Y, Mons U, Holleczek B, Saum KU, Brenner H. Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin Epigenetics. 2016; 8: 64. [CrossRef]
- Horvath S, Ritz BR. Increased epigenetic age and granulocyte counts in the blood of Parkinson's disease patients. Aging (Albany NY). 2015; 7: 1130-1142. [CrossRef]
- Marioni RE, Harris SE, Shah S, McRae AF, von Zglinicki T, Martin-Ruiz C, et al. The epigenetic clock and telomere length are independently associated with chronological age and mortality. Int J Epidemiol. 2016. [CrossRef]
- Lowe D, Horvath S, Raj K. Epigenetic clock analyses of cellular senescence and ageing. Oncotarget. 2016; 7: 8524-8531. [CrossRef]
- Lu AT, Xue L, Salfati EL, Chen BH, Ferrucci L, Levy D, et al. GWAS of epigenetic aging rates in blood reveals a critical role for TERT. Nat Commun. 2018; 9: 387. [CrossRef]
- Horvath S, Levine AJ. HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis. 2015; 212: 1563-1573. [CrossRef]
- Chen BH, Carty CL, Kimura M, Kark JD, Chen W, Li S, et al. Leukocyte telomere length, T cell composition and DNA methylation age. Aging (Albany NY). 2017; 9: 1983-1995. [CrossRef]
- Gensollen T, Iyer SS, Kasper DL, Blumberg RS. How colonization by microbiota in early life shapes the immune system. Science. 2016; 352: 539-544. [CrossRef]
- O’Callaghan NJ, Fenech M. A quantitative PCR method for measuring absolute telomere length. Biol Proced Online. 2011; 13: 3. [CrossRef]
- Montpetit AJ, Alhareeri AA, Montpetit M, Starkweather AR, Elmore LW, Filler K, et al. Telomere length: a review of methods for measurement. Nurs Res. 2014; 63: 289-299. [CrossRef]
- Aviv A, Hunt SC, Lin J, Cao X, Kimura M, Blackburn E. Impartial comparative analysis of measurement of leukocyte telomere length/DNA content by Southern blots and qPCR. Nucleic Acids Res. 2011; 39: e134. [CrossRef]
- Britt-Compton B, Rowson J, Locke M, Mackenzie I, Kipling D, Baird DM. Structural stability and chromosome-specific telomere length is governed by cis-acting determinants in humans. Hum Mol Genet. 2006; 15: 725-733. [CrossRef]
- Martin-Ruiz CM, Baird D, Roger L, Boukamp P, Krunic D, Cawthon R, et al. Reproducibility of telomere length assessment: an international collaborative study. Int J Epidemiol. 2015; 44: 1673-1683. [CrossRef]
- Wang F, Pan X, Kalmbach K, Seth-Smith ML, Ye X, Antumes DM, et al. Robust measurement of telomere length in single cells. Proc Natl Acad Sci U S A. 2013; 110: E1906-1912. [CrossRef]
- Mill J, Heijmans BT. From promises to practical strategies in epigenetic epidemiology. Nat Rev Genet. 2013; 14: 585-594. [CrossRef]
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012; 13: 86. [CrossRef]
- Burgess S, Timpson NJ, Ebrahim S, Davey Smith G. Mendelian randomization: where are we now and where are we going? Int J Epidemiol. 2015; 44: 379-388. [CrossRef]
- Gardner M, Bann D, Wiley L, Cooper R, Hardy R, Nitsch D, et al. Gender and telomere length: systematic review and meta-analysis. Exp Gerontol. 2014; 51: 15-27. [CrossRef]
