

New Insights into the Epigenetic Activities of Natural Compounds
Melita Vidakovic 1, †![]() , Jessica Marinello 2, †
, Jessica Marinello 2, †![]() , Maija Lahtela-Kakkonen 3
, Maija Lahtela-Kakkonen 3![]() , Daumantas Matulis 4
, Daumantas Matulis 4![]() , Vaida Linkuvienė 4
, Vaida Linkuvienė 4![]() , Benoît Y. Michel 5, †
, Benoît Y. Michel 5, †![]() , Ruta Navakauskienė 6
, Ruta Navakauskienė 6![]() , Michael S. Christodoulou 7, †
, Michael S. Christodoulou 7, †![]() , Danielle Passarella 8
, Danielle Passarella 8![]() , Saulius Klimasauskas 9
, Saulius Klimasauskas 9![]() , Christophe Blanquart 10
, Christophe Blanquart 10![]() , Muriel Cuendet 11
, Muriel Cuendet 11![]() , Judit Ovadi 12
, Judit Ovadi 12![]() , Stéphane Poulain 13, †
, Stéphane Poulain 13, †![]() , Fabien Fontaine-Vive 5
, Fabien Fontaine-Vive 5![]() , Alain Burger 5
, Alain Burger 5![]() , Nadine Martinet 5,*
, Nadine Martinet 5,*![]()
- Department of Molecular Biology, University of Beograd, Bulevar despota Stefana 142, 11000 Beograd, Serbia
- Department of Pharmacy and Biotechnology, University of Bologna, via Slemi 3, 40126 Bologna, Italy
- School of Pharmacy, University of Eastern Finland, ScP. O. Box 1627, 70211 Kuopio, Finland
- Department of Bio thermodynamics and Drug Design, Institute of Biotechnology, Vilnius University Graičiūno 8, LT-02241 Vilnius, Lithuania
- Institut de Chimie de Nice, Université Côte d’Azur and CNRS UMR 7272, Parc Valrose, Nice 06108 cedex, France
- Department of Molecular Cell Biology, Institute of Biochemistry, Life Sciences Center, Vilnius University, Sauletekio av. 7, LT-10257 Vilnius, Lithuania
- Dipartimento di Scienze Farmaceutiche, Università degli Studi di Milano, Via Venezian 21, 20133 Milano, Italy
- Dipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20133 Milano, Italy
- Department of Biological DNA Modification, Institute of Biotechnology, Vilnius University, Graičiūno 8, LT-02241 Vilnius, Lithuania
- CRCINA, INSERM, Université d’Angers et de Nantes, Nantes, 8 quai moncousu, BP7021 France
- School of pharmaceutical sciences, Université de Genève, Université de Lausanne, 1, Rue Michel-Servet, CH-1211, Geneva, Switzerland
- Institute of Enzymology, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar tudosok krt 2, Budapest, H-1117 Hungary
- Center for Biological Resources, Nancy, France, present address: Laboratory for Single Cell Technologies, Division of Genomic Medicine, RIKEN Center for Integrative Medical Sciences (IMS) 1-7-22, Suehiro-cho, Tsurumi-ku, Yokohama, 230-0045 Japan
† These authors contributed equally to this work.
* Correspondence: Nadine Martinet![]()
Received: May 30, 2018 | Accepted: July 30, 2018 | Published: August 16, 2018
OBM Genetics 2018, Volume 2, Issue 3 doi:10.21926/obm.genet.1803029
Academic Editors: Stéphane Viville and Marcel Mannens
Special Issue: Epigenetic Mechanisms in Health and Disease
Recommended citation: Vidakovic M, Marinello J, Kakkonen ML, Matulis D, Linkuvienė V, Michel BY, Navakauskienė R, Christodoulou MS, Passarella D, Klimasauskas S, Blanquart C, Cuendet M, Ovadi J, Poulain S, Vive FF, Burger AMartinet N. New Insights into the Epigenetic Activities of Natural Compounds. OBM Genetics 2018;2(3):029; doi:10.21926/obm.genet.1803029.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Background: With their varied pharmacophores, natural products are interesting tools to open the drug discovery pipeline. Several plant secondary metabolites are components of the human diet and have reported epigenetic activities. In this study, we screened a small natural compound library for epigenetic activities. Methods: Seventy-one different natural products plus 17 controls collected from all collaborating laboratories were screened. Localized DNA methylation (DNAm) was studied on a stretch of the retinoic acid receptor gene RARβ. All genomic 5-methylated cytosine (5mC) bases were then detected by high performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS). DNA methyl transferase 1 (DNMT1) enzymatic activity was measured for selected compounds. Level of histone H3 trimethylation at lysine 9 and 27 (me3H3K9 and me3H3K27) was measured by Western blot analysis. Global histone deacetylase inhibition (HDAC) was assayed first using a bioluminescent resonance energy transfer-based (BRET) assay and then with enzymatic fluorescence based-assays for most HDAC class 1. HDAC6 inhibition was measured by Western blot analysis. Sirtuin (Sirt2) inhibition was assessed first with a thermal shift assay and then using the enzymatic SIRTainty™ Class III HDAC assay for Sirt1 and Sirt2. Results: Diosmetin, (S)-equol, umbelliferone, papaverin and L-carnitine were identified as novel DNA demethylating agents. Emodin, rhein, aloin and D-glucuronic acid were identified as novel histone H3 demethylating compounds. Previously undescribed Sirt activation by apigenin, biochanin B, robinin, pinocembrin, aureusidine, brucine and boldine was also detected. Conclusions: High-throughput alpha screens are used for initial studies of diverse compound libraries; however, this approach has significant disadvantages for the study of DNAm. Indeed, finding unmethylated RARβ alleles in one cell line does not indicate the activity of the compound at the level of the entire genome over a given time-frame and a given dose. Measurement of DNMT1 activity is not useful since most natural compounds are not direct enzymatic inhibitors. When studying histone methylation, Western blot analysis is laborious but remains a cheap and effective assay under circumstances in which several histone methylases (KDMs) or demethylases may be responsible for modulation of histone methylation. Reversible epigenetic modifications of the genome remain feasible targets for nutrition-based preventive strategies. However, more accurate HDAC inhibition assays are still required for the evaluation of flavanols, which have fluorogenic properties that disturb classical fluorescence-based assays.
Keywords
DNA and histone H3 methylation; HDAC inhibition; natural products
1. Introduction
Epigenetics is defined as the heritable changes in a genome that occur without a change in the DNA nucleotide sequence. Since epigenetic modifications are reversible, they have been an important field of research for medicinal chemists. Several natural nutrients have been found to affect the epigenome through modification of DNA and histone methylation and histone acetylation as well as modifications of micro and long non-coding RNAs. We previously reported that several dietary polyphenols that are well-known as in vitro anti-oxidants are also documented epigenetic modifiers [1,2]; however, conflicting results were obtained [1,2,3,4]. Recent evidence supports a reduction in mortality risk associated with a polyphenol-rich diet [5]. In this study, we screened for epigenetic activities a small natural compound library assembled by a network of academic experts (COST action CIM1106). New positive results were obtained for each class of epigenetic activity studied. However, the limitations of fluorescence-based screening of some compounds were highlighted.
2. Materials and Methods
2.1 Reagents and Cell Culture
Reagents: Cell culture reagents, enzymes and compounds were obtained from Sigma–Aldrich (Saint Quentin en Yvelines, France) unless specified otherwise. Compounds sent by the authors were prepared for screening in the Institute of Chemistry in Nice (France). The compounds were first diluted to 10 mM in DMSO and 100 µL aliquots were stored in the dark and frozen at -20°C until shipment to each screening laboratory. The final working concentration of the compound prepared in cell culture medium was 15 µM, except for cell viability studies, HDAC screening and control compounds, for which several dilutions from 5 to 15 µM were assayed.
Cell culture: The MCF-7 (breast carcinoma) cell line was obtained from the American Type Culture Collection. Cells were cultured at 37°C in a humidified atmosphere of CO2/air (5%/95%). To prepare DNA for methylation studies, MCF7 cells were cultured in 2 triplicates in 24-wells plates seeded at a density of 6 × 104 cells/well. Cells were cultured for 5 days and the culture medium was replaced every other day (RPMI medium supplemented with 100 µg/mL streptomycin, 100 IU/mL penicillin and 10% v/v heat-inactivated fetal calf serum). After one day of culture, compounds (15µM) were added to wells and cultures lasted 4 more days. Subsequently, the cells were removed from wells into phosphate buffer saline using a rubber scraper and pelleted by centrifugation at 1000xg. The pellet was incubated for 12 h at 55°C in 1 mL lysis buffer (5 mM NaCl, 10 mM Tris-HCl pH 8, 0, 25 mM EDTA), 5 µL Triton X 100 and 5 µL proteinase K aqueous solution (20 mg/ml). Finally, DNA was purified using a classical phenol/chloroform/isoamyl alcohol extraction method. A working solution of each compound was prepared at 1 µg/µL in molecular biology grade water.
Cell viability: After 48 h in culture, cell viability was evaluated using the Cell Titer 96® assay kit (Promega, USA) according to the manufacturer’s recommendations. Results were expressed as IC50 values; i.e., the concentration of the compound inducing a 50% reduction in cell number by comparison with untreated control cultures containing 0.01% DMSO.
2.2 DNA Methylation (DNAm) Studies
DNA bisulfitation and methylation specific qPCR (MS-PCR): Purified DNA (100 ng) was prepared for MS-PCR using the EZ DNA Methylation™ Kit (ZYMO Research, USA) according to the manufacturer’s instructions. The methylation status of the RARβ second exon was determined by MS-PCR. Amplicon melting curves were generated using the Roche qPCR SYBR green kit according to the manufacturer's instructions, with annealing of the RARβ sense (GAGTAGGGTTTGTTTGGGTAT) and RARβ antisense (CCAAATAATCATTTACCATTTTCCA) primers at 57°C for 40 cycles. MS-PCRs were conducted in duplicate with a light cycler apparatus from Roche and interpreted using its allele-calling software version 2.0. Positive control DNA (0.01% DMSO-treated cells) served to distinguish fully methylated alleles (reported as 0.200 in Table 1), while negative control DNA (0.13 µM 5-azacytidine-treated cells reported as -0.250 in Table 1) served to distinguish fully unmethylated alleles; these results defined the extremities of a scale of methylation in which values > -0.100 were considered positive (DNA demethylation) because the qPCR allele-calling software was unable to resolve partially methylated alleles below this value.
Global DNAm study: Selected DNAs were also evaluated for global 5mC levels by HPLC-MS/MS. DNA was first treated with RNase-A to remove any contaminating RNA and then enzymatically digested to individual nucleosides for HPLC-MS/MS analysis as previously reported [6].
Table 1 shows the mean values of duplicate measurements expressed as the percentage of the ratio: 5mC/total cytosine. Fully methylated positive control RARβ alleles (0.01% DMSO treated) had a value of 1.70±0.36% and fully unmethylated negative control RARβ alleles (5-Aza-cytidine-treated) had a value of 0.88±0.09%. The 5-mC DNA ELISA™ (ZYMO Research, USA) was also used according to the manufacturer's instructions.
DNMT1 activity: Untreated MCF7 cells were harvested to prepare nuclear extracts using the Epiquik Nuclear Extraction Kit (Epigentek Inc. USA) following the manufacturer’s instructions. DNMT1 activity in nuclear extracts was determined using the Epiquik DNMT1 Activity Assay Kit (Epigentek Inc. USA) following the manufacturer’s protocol. DNMT1 activity in nuclear extracts was calculated as a percentage of that in the positive control (no compound).
In silico modeling: The human DNMT1 tridimensional structure was retrieved from the Protein Data Bank (3PTA code) and the Auto Dock Vina software [7] was used for in silico modeling of the compounds/enzyme interactions. The ligand-binding preference was determined by selecting the complexed structures with the binding lowest free energy among the different complexes predicted by docking calculations. The residues located at the S-adenosyl-methionine (SAM) cofactor site were then selected to conform to the docking boxes containing the catalytic and SAM sites; the results are presented in Supplementary Figure S1 and S2 for neoruscogenin and curcumin, respectively.
2.3 Histone H3K9 and K27 Studies
Western blot analysis was used to identify small molecules with the ability to decrease me3H3K9 and/or me3H3K27 levels in MCF7 cells treated as described previously [8]. Briefly, cells were cultured for 48 h as described in Section 2.1 and nuclear proteins were then investigated by Western blot analysis using antibodies for the specific detection of me3H3K9 and me3H3K27. GAPDH served as the protein loading control. The antibodies used were: monoclonal anti-GAPDH (ab8245, from Abcam, Cambridge, UK used at 1/100 dilution; polyclonal anti-me3H3K27 (No. 07-449) used at 1/300 dilution and polyclonal anti-me3H3K9 (No. 07-442, from Millipore) used at 1/200 dilution, and the protein bands were revealed with the horse radish peroxidase-conjugated goat anti-rabbit (or anti-mouse) secondary antibodies (1/200 dilution) from Dako Cytomation A/S (Copenhagen, Denmark). The intensity of immunoreactive protein bands was quantified by densitometry using the ImageJ 1.45 S software. Changes in the levels of me3HK27 and me3H3K9 were expressed as a percentage of the levels in the positive controls (MCF7 cells treated with BIX-01294 and with 3-deazaneplanocin A (DZNep); untreated cells served as the negative control. The catalytic activity of the KDM G9 was measured for (S)-evodiamine only with the EpiQuik™ KDM/Inhibition Assay (Epigentek, USA) as previously reported [8].
2.4 Histone H3K9 and K27 Studies
BRET: A previously designed high throughput BRET assay was used to test each compound (see for the Supplementary Figure S1 and S2) [9,10]. This assay is based on the use of engineered cells harboring two expression vectors; one encoding a bromo-domain, which recognizes acetylated histones, fused to Renilla luciferase (Rluc) and the second encoding histone H3 fused to the yellow fluorescent protein (YFP). If acetylated, histone H3-YFP recruits BrD-Rluc, and in the presence of the substrate of Rluc: coelenterazine, their proximity creates the conditions for an energy transfer between Rluc and YFP, which leads to fluorescent emission at 530 nm. Thus, this signal reflects the H3 acetylation created by an HDAC inhibitor. Toxicity of the compound was determined as the luciferase activity measured at 485 nm. Results were calculated as the mean values of three independent experiments at three different concentrations, with increased BRET signals indicating inhibitory activity. TSA, SAHA and CI994 served as positive controls.
General HDAC inhibitory activity: the FLUOR DE LYS™ HDAC fluorometric activity assay kit (Enzo Life Sciences, Lausen, Switzerland) was used according to the manufacturer’s recommendations. The provided HeLa nuclear extract served as the source of HDACs. Results were calculated as the mean of three independent experiments and expressed as IC50; SAHA was used as positive control [10,11].
Enzymatic assays: Selected compounds with an inhibitory activity ≥50 % were screened for HDAC1, 2 and 3 activities using a previously described method [10,11].
Western blot analysis of HDAC6: Acetylated tubulin levels were investigated in HeLa cells treated with selected compounds by Western blot analysis using an anti-tubulin antibody specific for K40 as previously described [10]. Tubastatin A served as the positive control.
Table 1 Screening results. In the compounds column, names are underlined when compounds were screened by Western blot analysis for HDAC6 inhibition as previously reported (11). In the biological activity column: IC50 values are indicated in (). The MS-PCR (methylation-specific PCR) scale ranges from -250 (fully unmethylated RARβ) to 250 (fully methylated RARβ). Sirt activation is denoted by a minus sign. DNMT: DNA methylation transferase; SAM: S-adenosyl-methionine; SAH: S-Adenosyl-L-homocysteine; BRET: Bioluminescent resonance energy transfer; HDAC: Histone deacetylase; Sirt: Sirtuin; me3H3K9 and me3H3K27: trimethylated histone H3 lysine 9 and 27.

2.5 Sirtuin Inhibition
Equilibrium binding ligands usually increase protein thermal stability by an amount proportional to the concentration and affinity of the ligand. This principle was used to develop a high-throughput screening assay for compounds with high binding affinity for human Sirt2. The thermal shift assay was performed using a Rotor Gene Q (Qiagen, Sydney, Australia) spectrofluorimeter. The Sirt2 protein concentration was 5 µM, the ligand concentration was 15 µM and the reaction volume was 10 µL. The samples were heated at a rate of 1°C/min and unfolding of the protein was monitored by measuring the fluorescence of 1, 8-anilinonaphthalene sulfonate (ANS) at 50–100 µM (excitation at 365 nm and emission at 460 nm). Data were analyzed as previously described [12]. The dissociation constant (Kd) of the control compound tenovin 6 was 15 µM at 37°C. Samples were analyzed in triplicate and values were expressed as the mean Kd with average variations <5%. Values >100 were considered to be significant. The Kd values for the control compounds cambinol and tenovin 6 were validated as previously described [13] using the FLUOR DE LYS® HDAC assay kit (Enzo Life Sciences, USA) according to the manufacturer’s instructions. This assay relies on the deacetylation of a fluorescently labeled acetylated peptide substrate (λExec = 360 nm and λEm = 460 nm). A nuclear magnetic resonance-based assay was also used [13].
Sirt enzymatic inhibition: Compounds were screened for modulation of Sirt1 and 2 activity using the SIRTainty™ Class III HDAC assay kit (Merck Millipore, USA) according to the manufacturer instructions (λExec = 420 nm and λEm = 450 nm). Modulation of Sirt activity was determined at 60 µM and data were presented as the mean weighted to the standard error; activation is denoted by a minus sign [14,15]. No humans, animals or plants were involved in this study.
3. Results
3.1 DNA Methylation Studies
Figure 1 shows the MS-PCR melting curves of the RARβ alleles. Fully methylated alleles (0.01% DMSO-treated cells) exhibited a peak at 77.5°C; while both unmethylated alleles (0.13 µM 5-azacytidine treated cells) exhibited a peak at 75.5°C. The results of all DNAm studies are shown in Table 1. Several compounds mediated strong demethylation of RARβ alleles under the culture conditions described; however, there were differences in the results of localized DNAm and global 5mC studies: diosmetin (-0.225 but no decrease by HPLC-MS/MS: 1.70 ± 0.36), genistein (-0.270 versus 1.12 ± 0.01), S-equol (-0.250 versus 1.15 ± 0.04), isoquercetine (-0.200 versus 1.12 ± 0.01), imperatorin (-0.250 versus 1.12 ± 0.01), umbelliferone (-0.170 versus 1.13 ± 0.01) and EGCG (-0.200 versus 1.12 ± 0.01), curcumin (-0.250 versus 1.09 ± 0.02), (S)-evodiamine (-0.180 versus 1.03 ± 0.02), and papaverin (-0.225 versus 1.10 ± 0.09). L-carnitine (-0.320 and 0.91 ± 0.02) exhibited the highest DNA demethylating activity. It was not possible to identify partially unmethylated RARβ alleles due to the lack of sensitivity of the allele-calling software. The HPLC-MS/MS assay is not suitable as a screening tool but its results corresponded relatively well with the results obtained using the 5-mC DNA ELISA™ (ZYMO Research, USA) according to the manufacturer's instructions previously [6,8]. For unmethylated alleles (0.13 µM 5-azacytidine treated cells), a 35% decrease in global 5mC content was detected by ELISA compared with a 48% decrease detected by HPLC-MS/MS.
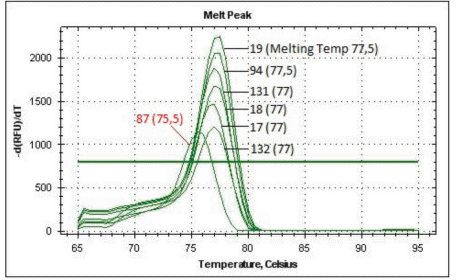
Figure 1 MS-PCR melting curves. DNA was extracted from MCF7 treated cells with 15 µM of each compound for 4 days. Bisulfite-treated DNA (100 ng) was analyzed by MS-PCR. 132 corresponds to 0.01% DMSO-treated MCF7 cells (fully methylated DNA, designated as +200 in Table 1) and 87 correspond to MCF7 cells treated with 0.13 µM 5 azacytidine (fully unmethylated control, designated as -250 in Table 1). 132: Resveratrol, 17: Vincamine, 18: Pilocarpine, 131: Tabersonine, 94: Protopine and 19: Cucurbitacine.
EGCG and curcumin also decreased the DNMT1 activity by 40% and approximately 70%, respectively.
In silico modeling indicated that most of the compounds did not bind to the DNMT1 catalytic site and were more likely to be SAH producers (Table 1). The results of molecular docking calculations showed that neoruscogenin and curcumin bind to the SAM pocket of DNMT1. The best binding modes corresponded to the highest binding affinity of the ligand in interaction with three amino-acids (MET 1169, GLY 1223 and ASN 1578) for neoruscogenin and with five amino-acids (GLU 1168, MET 1169, TRP 1170, ASP 1190 and ASN 1578) for curcumin; all amino-acids being located within 3.5Å around each ligand. The molecular modeling of these interactions is illustrated in Figure S1 and S2.
After 5 days of culture, several control compounds also produced marked decreases in 5mC levels. SGI-1027 is a commercially available inhibitor of DNMT1, DNMT3A and DNMT3B, which degrades the enzymes and competes with their cofactor, SAM. Chaetocin inhibits several KDMs such as SUV39 and G9a and causes a reduction in me3H3K9 via a SAM competitive pathway. DZNep acts as a SAM hydrolase inhibitor to induce the accumulation of SAH and also functions as a KDM EZH2 inhibitor. BIX-01294 is a G9a-like protein and G9a KDM inhibitor that does not compete with SAM [8]. Entinostat, valproic acid, anacardic acid and tenovin 6 have localized DNA demethylating activities that cannot be explained by SAM- or SAH-based mechanisms (Table 1). DNA and histone methylation studies of MCF7 cells were useful to investigate the roles of SAM and SAH in these processes.
3.2 Histone H3 Lysine 9 and 27 Trimethylation
Molecules with the ability to decrease me3H3K9 and/or K27 levels in MCF7 treated cells were identified by Western blot analysis; DZNep and BIX-01294 served as positive controls [8]. (S)-Evodiamine was identified as the most potent demethylating agent and induced decreased levels of me3H3K9 and me3H3K27 by 100% and 90%, respectively. All anthraquinones (emodin, rhein and aloin) were also active but exhibited greater activity for me3H3K9 compared with that for me3H3K27, while D-glucuronic acid was almost as efficient as (S)-evodiamine. G9 exhibited 30% lower activity with (S)-evodiamine and 80% lower activity with BIX-01294 and DZNep. We reported previously that (S)-evodiamine exhibited high pharmacophoric similarity with SAM and suggested that this was the basis for its ability to inhibit G9 activity [8]. Emodin, rhein and glucuronic acid did not exhibit DNA demethylating activity, suggesting a KDM activity not based on SAM/SAH modulation mechanisms. However, the mechanism underlying the direct inhibition of G9 remains to be confirmed.
3.3 HDAC Inhibition
We previously conducted a comprehensive comparison of our BRET screen efficiency with that of the nuclear global HDAC assay [9,10,11]. The BRET assay was performed on living cells and at compound concentrations of 100, 50, 25 and 12.5 µM. This assay was very sensitive to the solubility of the assayed compound and also to its potential toxicity. Apigenin, galangin, scutallerein, kaempferol, ermanin, robinin, morin, myricitrin, quercetin, isoquercetin, rutin, epicatechin, catechin, chlorogenic acid, emodin, rhein and aloin were found to have significant global HDAC inhibitory activity in both assays. However, galangin, ermanin, robinin, morin, myricitrin, quercetin, isoquercetin, rutin, emodin, rhein and aloin also have emissive capacities (excitation λExec) = 340–360 nm and emission (λEm) = 440–465 nm) with the potential to interfere with further enzymatic assays; thus, only the non-fluorogenic compounds, apigenin and scutallerein, were further analyzed. These compounds can be considered as true pan-HDAC inhibitors that inhibit HDAC3 with high efficiency (IC50: 31.2 ± 10.6 µM and 18.0 ±7.9 µM, respectively). None of the tested compounds demonstrated HDAC6 inhibitory activity in Western blot analyses [11]. Similar results have been reported for the same compounds (flavones and flavanols) previously [16], although the potential emissive properties of the screened compounds was not addressed despite being recently documented as a concern [17].
3.4 Sirtuin Modulation
The results of sirtuin modulation assays are shown in Table 1. The thermal shift assay is a good indicator of the fluorogenic activity of most flavonoids since most absorb between 330–380 nm; these are indicated as “Fluo” in Table 1. For example: biochanin A absorbs at 338 nm and emits at 488 nm. However, flavonoids do not exhibit such emissive capacity in the SIRTainty™ assay. Thus, the weak Sirt inhibition observed for apigenin (Sirt2: 32.8 ± 1.2) and the activation of Sirt1 by biochanin B (-14.2 %) detected using this kit can be considered to be reliable results. The same consideration applies also to the inhibition of both Sirt1 and Sirt2 by quercetin (33.6 ± 1.2 and 52.7 ± 1.6, respectively) and curcumin (10.7 ± 0.4 and 40.2 ± 12.8, respectively). The relatively low Sirt1 activation by pinocembrin (approximately -10%) is also reliable. This is also the case for aureusidine, which has a high emissive capacity in the HDAC enzymatic assays but not in the SIRTainty™ assay. Aureusidine was found to be a strong Sirt1 activator (-25.6 ± 5.8 higher than for the reference compound resveratrol). When testing alkaloids, we did not observe any such emissive behavior and (S)-evodiamine mediated potent Sirt inhibition (Sirt1: 42 ± 3.6, Sirt2: 71 ± 2.1 and Sirt3: 20 ± 2.1), which is consistent with our previous report [18]. Brucine induced obvious activation of Sirt1 (-10.5 ± 0.1) and potent inhibition of Sirt2 (74.9 ± 16). Boldin induced very weak activation of Sirt1 (-4.1 ± 1) and weak inhibition of Sirt2 (24.62 ± 6.65). Among the terpenoides, neoruscogenin mediated weak inhibition of both Sirt1 and Sirt2 (21.62 ± 0 and 26.5 ± 2.3, respectively), while cycloartenol mediated weak activation of Sirt1 alone (-4.4 ± 1). The anthraquinones were highly fluorogenic in all enzymatic HDAC assays, with the exception of the Sirt inhibition mediated by aloin (Sirt1: 39 ± 2.2 and Sirt2: 39.7 ± 6.9). Inhibition of Sirt6 was also recently reported for kaempferol, quercetin and myricetin [19].
3.5 Cell Viability
Most of the assayed compounds had very little effect on cell viability. The lowest IC50 was detected for apigenin (3 µM), genkwanin (9 µM) and several of the alkaloids including (S)-evodiamine, papaverin and brucine (0.9 µM). We used these IC50 values only to optimize our cell cultures conditions for control compounds such as 5-azacytidine. All values are reported in Table 1.
4. Discussion
Given the widespread implications of the diet in the regulation of the epigenome, a strong evidence-based knowledge of the effects of food components is required. We have previously reviewed the epigenetic activities of various dietary components [1,2] and in this study, we screened a small natural compound library for epigenetic activities. Some plants in which these compounds can be found are listed in Table 1. We identified previously unreported DNA methylation activities of diosmetin, S-equol, umbelliferone, papaverin and L-carnitine as well as histone methylation activity of emodin, rhein, aloin and D-glucuronic acid (1, 2). Finally, we identified previously unreported Sirt activation by apigenin, biochanin B, robinin, pinocembrin, aureusidine, brucine and boldine.
Phytochemicals can be classified into major categories, such as polyphenols, including phenolic acids, flavonoids, and stilbenes/lignans. Flavonoids can be further divided into different groups such as, flavones, flavanones, isoflavones, and flavanols, based on similar chemical structures. Thus, it can be speculated that compounds with a given structure will be associated with similar functions. The results of the present study indicate that most flavanols have HDAC inhibition properties although this conclusion must be tempered by the knowledge that most phyto-nutrients undergo intestinal transformation mediated by microbiota and enterocyte enzymes prior to absorption. Flavonoids are known to be poorly absorbed and most of what is absorbed, exists as metabolites that are further modified for rapid excretion [20]. The flavanols tested in this study are certainly not the final forms found in plasma. Their complex metabolic fate prevents an easy understanding of their potential health effects. In addition, their in vitro and in vivo activities do not necessarily correlate, a phenomenon that is best exemplified by the fact that dietary polyphenols are active in vitro but have little or no direct antioxidant value following digestion [20]. In addition, evaluation of the activity of many phytochemicals is further complicated by the fluorogenic potential of many of these compounds. Thus, the epigenetic bioactivities of phyto-nutrients remain to be elucidated definitively, even when using laborious methods similar to those used in this study. Indeed, selection of the optimal conditions and cell lines for screening assays is a significant challenge.
Aberrantly methylated DNA can be identified on a gene per gene basis or using various higher throughput technologies; however, the costs involved and the interpretation of results are not the same. A recent epidemiological study was conducted to evaluate the in vivo effects of green tea consumption on human DNA. The analysis revealed that 28 regions were differentially methylated in relation to green tea consumption, but only in women [21]. This indicates that some genomic regions are relatively more sensitive to DNA demethylating compounds, which are known to act through several mechanisms. Catechol-containing compound such as (+)-catechin decreases DNA methylation indirectly by consuming SAM (SAM pumping) for their own methylation through cathechol-O-methyltransferase-mediated O-methylation. This process leads to excessive SAH production, which inhibits DNMT1 directly. Alternatively, some compounds prevent the methylation of the newly synthesized DNA strand by occupation of the catalytic pocket of DNMT1, while others bind to the DNMT1 active site and generate H2O2, which is deleterious to the enzyme. In addition, some compounds prevent DNA methylation by competing with the DNMT1 cofactor, SAM. The first three mechanisms were reported to apply to EGCG, which is the most abundant catechin in green tea [1,2]. DNMT1 inhibition by EGCG requires an intact gallic acid moiety, although EGCG is often rearranged to reactive quinones, losing the necessary gallic acid moiety. Moreover, in plasma and after glucuronidation, EGCG has very poor bioavailability. Other tea polyphenols, such as (+)-catechin, (-)-epicatechin and quercetin have been reported to have similar, but weaker epigenetic activities [1,2]. Under in vitro conditions, curcumin is poorly soluble in water and is mainly found in its enol form, acting as a Michael acceptor to covalently block the catalytic DNMT1 thiolate C1226. In vivo, curcumin is metabolized as a glucuronide and rapidly degraded at alkaline pH [1]. In silico, curcumin binds the DNMT1 catalytic site (Figure S2). The major isoflavones from soybean, genistein, and biochanin A, have also been reported to have DNA demethylating activity [1,2,3,4]. Although their mechanisms of action remain to be fully elucidated, in this study, we show that S-equol, an isoflavone metabolite, remains a strong DNA demethylating agent. Most flavanols acted as pan-HDAC class 1 inhibitors although, as previously emphasized, further specific screening is required because these compounds also have emissive capacities capable of interfering in fluorescence-based assays. Anthraquinones have Sirt-modulating capacities and also induced marked decreases in me3H3K9 and K27 levels, although the mechanisms of action require further investigation. Alkaloids are a rich source of drugs, among which brucine is a strong Sirt1 activator.
5. Conclusions
High-throughput screening is a well-established starting point for most discovery research projects conducted by medicinal chemists, although the appropriate endpoints for epigenetic testing including individual enzymes, histones or/and non-histones proteins modified by the enzymes, remain to be clarified. Robust assays to identify compounds, or rather metabolites of compounds, that modulate epigenetic markers in cells are required. These assays must effectively avoid false positive results and provide information that can be translated for the development of evidence-based preventive epigenetic diets. Based on these requirements, we believe that the epigenetic activity of phyto-nutrients cannot yet be conclusively determined using the currently available techniques. Indeed, the fluorogenic properties of compounds should be screened systematically before further testing.
Acknowledgments
This research has been developed under the umbrella of CM1106 COST Action “Chemical Approaches for Targeting Drug Resistance in Cancer Stem Cells”. Thanks are also addressed to the COST Actions CM1406 (EPICHEM) and OTKA K-11214. The authors thank Zita Liutkevičiūtė for HPLC/MS analysis of modified nucleosides.
Additional Materials
The following additional materials are uploaded at the page of this paper.
1. Figure S1: Neoruscogenin docking into DNMT1.
2. Figure S2 : Curcumin docking into DNMT1.
Author Contributions
Melita Vidakovic conducted experiments and analyzed the results: cell viability studies, DNMT1 enzymatic measure, DNA conditioning and part of the DNAm and Sirt study. Jessica Marinello realized most of the MS-PCR. Maija Lahtela-Kakkonen was responsible for most of the Sirt enzymatic screening. Daumantas Matulis and Vaida Linkuvienė were responsible for the Sirt2 alpha screen by thermal shift. Benoit Michel, Stéphane Poulain and Michael. S Christodoulou contributed to the interpretation and analysis of the data. Alain Burger and Danielle Passarella contributed to the writing of the manuscript. Fabien Fontaine-Vive realized the DNMT1 in silico modelling study. Ruta Navakauskienė was responsible for the me3H3K9 and 27 Western blots. Saulius Klimasauskas was responsible for the HPLC-MS/MS measure of 5mC levels. Christophe Blanquart was responsible for the BRET screening. Muriel Cuendet was responsible for the HDAC second and third screening. Judit Ovadi was in charge of the HDAC6 screening by Western blot. Nadine Martinet conceived and coordinated the experimental efforts, and wrote this paper which was approved by all authors.
Funding
Melita Vidakovic was the recipient of the exchange program PAVEL SAVIC from the French and the Serbian Ministries of foreign affairs. This work was also supported by INSERM, CNRS, Région Pays de la Loire, the ‘Institut de recherche en santé respiratoire des Pays de la Loire’ and the ‘Ligue Contre le Cancer (committees of Morbihan, Sarthe, Vendée et Loire-Atlantique) : ARSMESO44. Ruta Navakauskienė is thankful for partial funding of this work by the Research Council of Lithuania (Project No. SEN-12/2015).
Competing Interests
The authors have declared that no competing interests exist.
References
- Marzag H, Warnault P, Martinet N, Bougrin K, Benhida R. Studies in Natural Products Chemistry, (Bioactive Natural Products). 1st ed. Amsterdam: Elsevier Science Publishers; Ed. Atta-ur-Rahman. 2014; 41: 191–226.
- Vidakovic M, Christodoulou MS, Thomas A, Poulain S, Lahtela-Kakkonen M, Matulis D, et al. Can we use the epigenetic bioactivity of caloric restriction and phytochemicals to promote healthy ageing?. Med. Chem. Commun. 2014; 5: 1804–1820. [CrossRef]
- Aggarwal R, Jha M, Shrivastava A, Jha AK. Natural Compounds: Role in Reversal of Epigenetic Changes. Biochemistry. 2015; 80: 972–989. [CrossRef]
- Zam W, Khadour A. Impact of Phytochemicals and Dietary Patterns on Epigenome and Cancer. Nutr. Cancer. 2017; 69: 184–200. [CrossRef]
- Pounis G, Costanzo S, Bonaccio M, Di Castelnuovo A, de Curtis A, Ruggiero E, et al. Reduced mortality risk by a polyphenol-rich diet: An analysis from the Moli-sani study. Nutrition. 2018; 48: 87–95. [CrossRef]
- Myrianthopoulos V, Cartron PF, Klimasauskas S, Matulis D, Bronner C, Martinet N, et al. Tandem virtual screening targeting the SRA domain of UHRF1 identifies a novel chemical tool modulating DNA methylation. Eur. J. Med. Chem. 2016; 114: 390–396. [CrossRef]
- Trott O, Olson AJ. Auto Dock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010; 31: 455–461.
- Navakauskienė R, Mori M, Christodoulou MS, Zentelytė A, Botta B, Dalla Via L, et al. Histone demethylating agents as potential S-Adenosyl-L-methionine-competitors. Med. Chem. Commun, 2016; 7: 1245–1255. [CrossRef]
- Blanquart C, Francois M, Charrier C, Bertrand P, Gregoire M. Pharmacological characterization of histone deacetylase inhibitor and tumor cell-growth inhibition properties of new benzofuranone compounds. Curr. Cancer Drug Targets. 2011; 11: 919–928. [CrossRef]
- Zwick V, Nurisso A, Simões-Pires C, Bouchet S, Martinet N, Lehotzky A, et al. Cross metathesis with Hydroxamate and Benzamide BOC-protected alkenes to access HDAC inhibitors and their biological evaluation highlighted intrinsic activity of BOC-protected dihydroxamates. Bioorg. Med. Chem. Lett. 2016; 26: 154–159. [CrossRef]
- Zwick VT, Chatzivasileiou A-O, Deschamps N, Roussaki M, Simoes-Pires CA, Nurisso A, et al. Aurones as histone deacetylase inhibitors: identification of key features. Bioorg. Med. Chem. Lett. 2014; 24: 5497–5501. [CrossRef]
- Zubriene A, Matuliene J, Baranauskiene L, Jachno J, Torresan J, Michailoviene V, et al. Measurement of Nano molar dissociation constants by titration calorimetry and thermal shift assay - radicicol binding to Hsp90 and ethoxzolamide binding to CAII. Int. J. Mol. Sci. 2009; 10: 2662–2680. [CrossRef]
- Pirrie L, McCarthy AR, Major LL, Morkūnaitė V, Zubrienė A, Matulis D, et al. Discovery and validation of SIRT2 inhibitors based on tenovin-6: use of a ¹H-NMR method to assess deacetylase activity. Molecules. 2012; 17: 12206–12224. [CrossRef]
- Kim E, Bisson WH, Löhr CV, Williams DE, Ho E, Dashwood RH, et al. Histone and Non-Histone Targets of Dietary Deacetylase Inhibitors. Curr. Top. Med. Chem. 2016; 16: 714–731. [CrossRef]
- Michan S, Sinclair D. Sirtuines in mammals: insights into their biological function. Biochem. J. 2007; 404: 1–13. [CrossRef]
- Godoy LD, Lucas JE, Bender AJ, Romanick SS, Ferguson BS. Targeting the epigenome: Screening bioactive compounds that regulate histone deacetylase activity. Mol. Nutr. Food Res. 2017; 61: doi: 10.1002/mnfr.201600744. [CrossRef]
- Wen H, Xue N, Wu F, He Y, Zhang G, Hu Z, et al. Exploration of the Fluorescent Properties and the Modulated Activities against Sirtuin Fluorogenic Assays of Chromenone-Derived Natural Products. Molecules. 2018; 23: E1063. [CrossRef]
- Christodoulou MS, Sacchetti A, Ronchetti V, Caufin S, Silvani A, Lesma G, et al. Quinazolinecarboline alkaloid evodiamine as scaffold for targeting topoisomerase I and sirtuins. Bioorg. Med. Chem. 2013; 21: 6920–6928. [CrossRef]
- Rahnasto-Rilla M, Tyni J, Huovinen M, Jarho E, Kulikowicz T, Ravichandran S, et al. Natural polyphenols as sirtuin 6 modulators. Bioavailability of the polyphenols: status and controversies. Sci. Rep. 2018; 8: 4163. [CrossRef]
- D'Archivio M, Filesi C, Varì R, Scazzocchio B, Masella R. Bioavailability of the polyphenols: status and controversies. Int. J. Mol. Sci. 2010; 11: 1321–1342. [CrossRef]
- Ek WE, Tobi EW, Ahsan M, Lampa E, Ponzi E, Kyrtopoulos SA, et al. Tea and coffee consumption in relation to DNA methylation in four European cohorts. Hum. Mol. Genet. 2017; 26: 3221–3231. [CrossRef]
