

Identification and Prioritization of Causal Variants of Human Genetic Disorders from Exome or Whole Genome Sequencing Data
Nagarajan Paramasivam 1,2,*![]() , Martin Granzow 3
, Martin Granzow 3![]() , Christina Evers 3
, Christina Evers 3![]() , Katrin Hinderhofer 3
, Katrin Hinderhofer 3![]() , Stefan Wiemann 4
, Stefan Wiemann 4![]() , Claus R. Bartram 3
, Claus R. Bartram 3![]() , Roland Eils 1,5,#
, Roland Eils 1,5,#![]() , Matthias Schlesner 1,6,*,#
, Matthias Schlesner 1,6,*,#![]()
- Division of Theoretical Bioinformatics (B080), German Cancer Research Center (DKFZ), Heidelberg, Germany
- Medical Faculty Heidelberg, Heidelberg University, Germany
- Institute of Human Genetics, University of Heidelberg, Heidelberg, Germany
- Genomics and Proteomics Core Facility, German Cancer Research Center (DKFZ), Heidelberg, Germany
- Department for Bioinformatics and Functional Genomics, Institute for Pharmacy and Molecular Biotechnology (IPMB) and BioQuant, Heidelberg University, Heidelberg, Germany
- Bioinformatics and Omics Data Analytics (B240), German Cancer Research Center (DKFZ), Heidelberg, Germany
#shared senior authorship
* Correspondence: Nagarajan Paramasivam![]() , Matthias Schlesner
, Matthias Schlesner![]()
Received: October 24, 2017 | Accepted: March 20, 2018 | Published: April 16, 2018
OBM Genetics 2018, volume 2, issue 2 doi:10.21926/obm.genet.1802017
Academic Editors: Ute Moog and Domenico Coviello
Special Issue: Next Generation Sequencing
Recommended citation: Paramasivam N, Granzow M, Evers C, Hinderhofer K, Wiemann S, Bartram CR, Eils R, Schlesner M. Identification and Prioritization of Causal Variants of Human Genetic Disorders from Exome or Whole Genome Sequencing Data. OBM Genetics 2018;2(2):017; doi:10.21926/obm.genet.1802017.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Background: With genome sequencing entering clinics as a diagnostic tool to detect genetic disorders, there is an increasing need for bioinformatics-based solutions that enable precise causal variant identification in a timely manner. Workflows for the identification of candidate disease-causing variants usually perform the following tasks: i) identification of variants; ii) filtering of variants to remove polymorphisms and technical artifacts; and iii) prioritization of remaining variants to provide a small set of candidates for further analysis.
Methods: Here, we present a pipeline designed to identify variants and genes from trio sequencing or pedigree-based sequencing data that prioritizes them into distinct hierarchical tiers.
Results: We applied this pipeline to a study of patients with neurodevelopmental disorders of unknown cause and identified causal variants in more than 35% of cases.
Conclusions: Classification and prioritization of large numbers of variants into different tiers can help to obtain a smaller set of candidates to facilitate downstream analysis for identification of causal variants of genetic diseases.
Keywords
NGS; trio-sequencing; rare genetic disorders; variant prioritization
1. Introduction
Next generation sequencing (NGS) has proven to be a powerful technique to identify causal genes of rare genetic disorders [1,2]. The recent decline in sequencing costs has enabled the use of NGS for diagnostics within the broader clinical setting [3,4,5]. However, more widespread generation of sequence data has created new challenges related to the daunting tasks of managing, analyzing, and interpreting large data sets [6]. Consequently, bioinformatics tools and pipelines must be constantly improved to enable more rapid and effective NGS data analysis to keep pace with the flood of data.
More than three million variants are present in individual genomes that distinguish them from the currently used human reference genome [7]. For clinical sequencing projects, this number of variants somehow must be reduced to a manageable number of candidate variants before conducting downstream analyses to identify causal variants of specific genetic diseases. One effective strategy to reduce the number of candidate variants has employed trio sequencing, whereby healthy parents are sequenced along with their affected children.
Generally, workflows for trio- or pedigree-based analysis share three basic steps [8]. In step one, raw sequence reads are mapped to the reference genome for each sample individually then variants for all samples are analyzed together and the genotype of each sample is determined. In step two, technical artifacts and variants which are common in the population are removed, as they are highly unlikely to cause rare genetic diseases. Even after this first filtering step, an unmanageable number of potential candidate variants still remains, requiring prioritization using pedigree information and various annotations to further enrich the list for causal variants of disease [8].
Here, we present a complete workflow for candidate variant identification and prioritization for analysis of whole exome sequencing (WES) and whole genome sequencing (WGS) data generated from trio sequencing or from larger pedigree analyses.
2. Material and Methods
We developed a workflow that incorporates read alignment, variant calling, annotation, filtering, and prioritization steps. A particular focus is placed on various filtering and annotation steps to prioritize variants depending on an assumed inheritance model for disease in each family before performing further analyses.
2.1. Read Alignment
Raw sequencing data were mapped to the 1000 Genomes Reference Genome Sequence (hs37d5) [7] using the Burrows-Wheeler Aligner (BWA) [9] aln algorithm (Version 0.6.2) with standard parameter settings except for the setting ‘-q 20’. The resulting SAM files were sorted, converted to BAM format and indexed using SAMtools-0.1.19 [10]. Multiple lanes per sample were merged and duplicate reads marked using Picard [11] with the following parameter settings, ‘picard-1.61 MarkDuplicates VALIDATION_STRINGENCY=SILENT REMOVE_DUPLICATES=FALSE ASSUME_SORTED=TRUE MAX_RECORDS_IN_RAM=12500000 CREATE_INDEX=TRUE CREATE_MD5_FILE=TRUE’.
2.2. Variant Calling and Annotation
Single nucleotide variants (SNVs) and small insertion-deletion variants (indels) of 1-20 bps were jointly called from all samples obtained from each family using Platypus [12] with following parameter settings, ‘Platypus_0.8.1.py callVariants nCPU=10 genIndels=1 genSNPs=1 minFlank=0 -bamFiles=\List_of_Bam_Files’ --refFile=hs37d5.fa --output=Output_VCF’. Gene and transcript definitions from Gencode v19 [13] were added using ANNOVAR [14] and minor allele frequency (MAF) information from 1000 Genomes Phase III and Exome Aggregation Consortium (ExAC) [15] databases was added. In addition, allele frequencies from 328 WES and 177 WGS inhouse samples (referred to as local controls) were assigned to the variants.
2.3. Variant Filtering
Variants that remained after employing all Platypus internal filters were considered further. Frequent variants were removed based on a MAF threshold of 0.1% from ExAC and 1000 Genomes Phase III databases. To remove technical artifacts specific to our pipeline, variants that were present in local controls above the threshold of 2% were considered to be artifacts and were removed.
Next, trio sequencing variant data from each patient and healthy parents were combined to efficiently reduce the number of candidates. Only variants fulfilling an assumed inheritance model for a given genetic disease were considered further. For an autosomal dominant (AD) disease, only de novo variants, i.e. variants which are present in the patient but not in any of the parents, were considered as candidates. In the case of autosomal recessive (AR) inheritance from consanguineous parents, we expected the genotype to be homozygous in patients and heterozygous in both parents. Hemizygous variants were assumed to be heterozygous in only one of the parents and homozygous in the patient. Candidates for X-linked (XL) variants were assumed to be heterozygous in the mother and hemizygous in the male patient. Finally, to identify candidates for compound heterozygous inheritance of AR diseases, SNVs and indels were combined to find two different heterozygous mutations within the same gene in the patient, with one variant from one parent and the other variant from the other parent.
Lastly, variants with low genotype quality (Phred score <20) in at least one sample from the family were filtered out. The remaining variants were prioritized to select a list of causal variant candidates for further downstream analysis.
2.4. Variant Prioritization
Both variants and genes were prioritized using separate analyses and then classified into different tiers from which the final candidates were selected. Variants were ultimately prioritized based on their effects on protein function, which were predicted from various conservation scores. To this end, various annotations from dbNSFP [16], including the GERP score [17] and CADD scores [18], were assigned to the variants. The GERP score [17] measures the evolutionary conservation of sequences across species; a position with a score greater than two is considered to be a highly conserved nucleotide and its alteration is likely to have a high functional impact. CADD scores [18] integrate various annotations including sequence conservation scores and ENCODE project functional annotations to measure the deleteriousness of a variant. A CADD score of 13 derived using the Phred scale was used as a threshold such that prioritized variants above that threshold were considered to be within the top 5% of deleterious variants within the human genome.
As illustrated in Figure 1, genes were also prioritized based on their intolerance towards functional mutations. Intolerance missense z or pLI scores from ExAC were added if the variant was a missense or loss-of-function (LoF) variant, respectively (LoF, includes stop gain/loss, splice acceptor/donor or frameshift indels). We considered genes with Z score >2 as intolerant to missense variants, while genes with pLI scores > 0.9 were considered intolerant to LoF variants.
Finally, variants were categorized into two tiers, one for LoF variants and one for missense variants and each tier contained three levels. Level 0 of both tiers contained the whole variant set before prioritization. Next, LoF variants were moved to level 1 of tier 1 then LoF variants with CADD score above the threshold were moved further into level 2. Finally, variants in level 2 which could affect gene function while also having pLI scores above threshold were moved into level 3. Meanwhile, missense variants were moved from level 0 into level 1 of tier 2 and further prioritization of variants into higher levels of tier 2 was done as for tier 1, except that instead of the ExAC pLI score, the ExAC missense Z score was used to prioritize variants into level 3. Initially, for downstream analysis only variants in level 3 of both tiers were considered. Only after no candidates were found in level 3 were variants in lower levels examined.
Ultimately, prioritized variants were further classified by medical geneticists using the guidelines provided by the American College of Medical Genetics and the Association for Molecular Pathology [19].
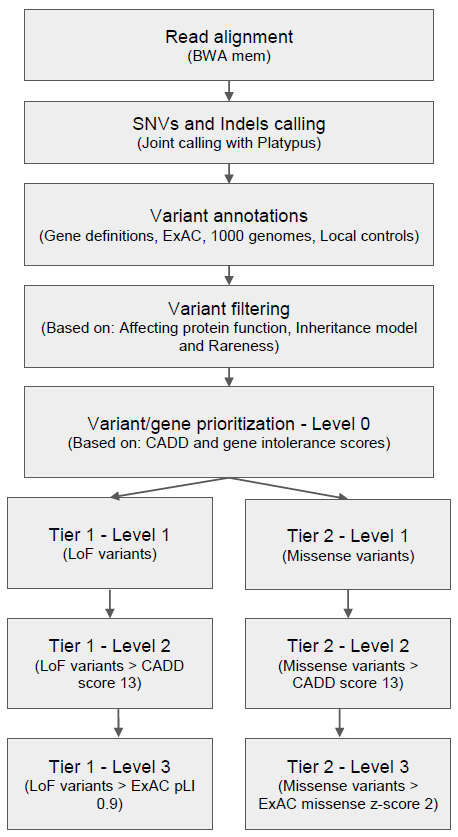
Figure 1 Germline variant analysis pipeline for rare genetic disorders.
3. Results
Recently we published results from our clinical exome initiative [4], where we analyzed exome data from 60 families with undiagnosed neurodevelopmental disorders (NDD), neurometabolic disorders (NMD), and dystonias and found causal variants in 35% of families. In this study, we used the results from 39 families within the NDD cohort to demonstrate how the germline analysis pipeline effectively filtered and prioritized variants identified from WES data obtained using a trio-based sequencing strategy.
Among the 127 WES samples from 39 families, 201,687 SNVs and 38,186 indels were present, with a minimum coverage of 10x. Among these variants, 152,641 SNVs and 14,912 indels present in ExAC or 1000 Genome Phase III databases with MAF values above 0.1% were discarded, as were variants detected in local controls with a frequency above 2%. After this step, 6,368 SNVs and 451 indels remained and were used for further analysis.
In the next step, all variants outside of exonic regions (+/- two base pairs to account for splice sites as defined by the Gencode v19 gene model) were removed. The remaining variants were classified according to their predicted effect on protein function. After variants that did not cause missense and LoF mutations were discarded, 528 SNVs and 29 indels remained which were considered rare/private variants with predicted protein functional effects.
For all 39 families of the NDD cohort, trio sequencing was performed. In addition, in some families, genomes of affected or unaffected siblings were also sequenced to facilitate variant filtering for recessive and X-linked diseases. For analysis, we considered such cases as separate trio families; only in the final steps of the pipeline were results merged, analyzed, and reported for each family unit. The pedigree information was used to classify variants into de novo, homozygous, hemizygous, and heterozygous variants (Figure 2). Heterozygous SNVs and indels were further combined to find compound heterozygous variants present in these families. At the end of the variant filtering steps, an average of four de novo, five homozygous, one hemizygous, and two pairs of compound heterozygous small variants remained per family. These variants were further prioritized into different tiers to select the best candidate causal variants/genes for further analysis and confirmation.
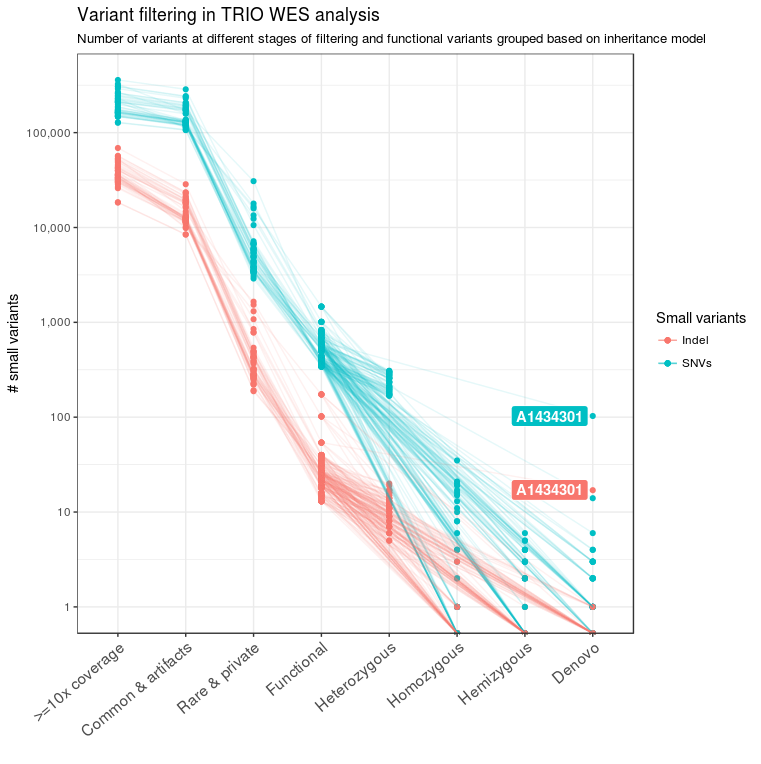
Figure 2 Variant filtering in TRIO WES analysis. The number of candidate variants after the different filtering steps is shown for the 39 families from the NDD cohort. After applying the basic quality filters, functional variants were identified and patient and parent data were combined. Based on pedigree information, functional variants were further classified into different genetic models. The sample A1434301 contained a high number of de novo variants due to poor DNA quality.
A total of 69 variants from all samples were annotated as LoF variants and were moved to level 1 of tier 1. Of those variants, 49 had CADD scores >13 and were moved to level 2 of tier 1; 12 of these 49 variants were detected in genes that were predicted to be intolerant to LoF variants and were thus moved to level 3 of tier 1 (Figure 3). A total of 529 missense variants were moved to level 1 of tier 2. Of these, 296 variants had CADD scores >13 and were moved to level 2 of tier 2; 67 of these 296 variants were mapped to genes with ExAC missense Z scores >2 and were moved to level 3 of tier 2 (Figure 4).
Ultimately, using these approach candidate causal variants could be identified in 15 families of the NDD cohort (table 3, [4]). The inheritance pattern was AD in six of these families, AR in seven families and XL in two families. Five of the AD causal variants and two XL causal variants were found in level 3 of tier 1 or 2, where the variants and genes satisfied all thresholds set in the prioritization step. Interestingly, only one of seven AR families had a variant in the top level of tier 2. The other AR causal variants had high variant deleteriousness scores, but the genes were predicted to be tolerant to new missense mutations. It should be noted that intolerance scores were not developed for AR inheritance models. Therefore, gene intolerance score-based prioritization should be relaxed in order to maximize the likelihood of finding causal variants of AR inherited diseases.
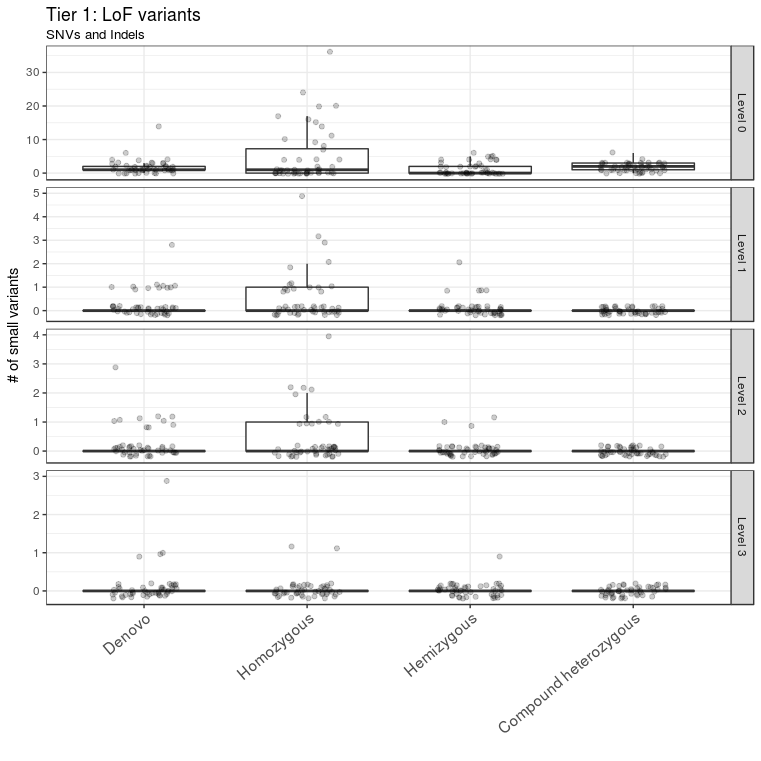
Figure 3 LoF variant prioritization. LoF variants were classified into different levels in tier 1. LoF variants with CADD score > 13 and ExAC pLI score > 0.9 were assigned to level 3. They constitute the first set of candidate variants to be considered for further downstream analysis. The outlier A1434301 is not shown.
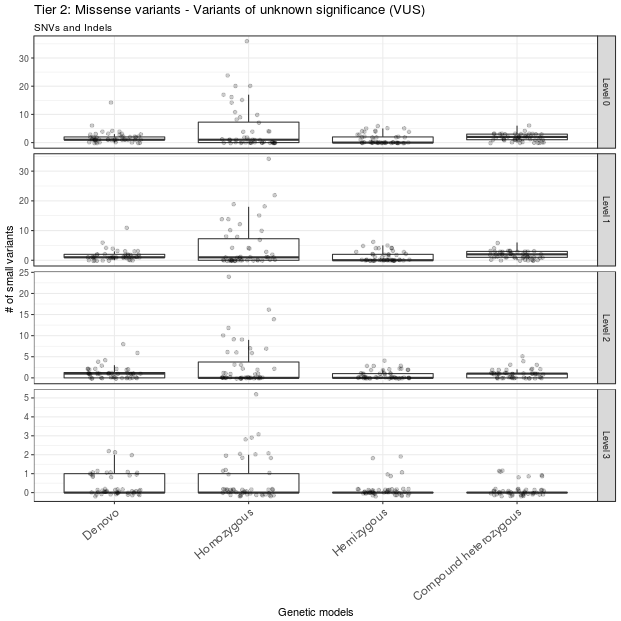

Figure 4 Missense variants prioritization. Missense variants were classified into different levels of tier 2. Only missense variants with CADD score > 13 and ExAC missense Z score > 2 are in level 3. These variants constitute the second set of variants to be considered after ruling out the level 3 variants in tier 1. The outlier A1434301 is not shown.
4. Discussion
Next-generation sequencing makes it possible to identify causal variants of genetic diseases without the need for prior hypothesis pinpointing a specific affected gene. Rapidly decreasing costs for NGS now permit the routine application of whole exome or even whole genome sequencing for use as clinical diagnostic tools. However, genome sequencing in the absence of other information generates long lists of candidate variants of uncertain relevance to disease. Moreover, the use of manual variant filtering and candidate selection based on background knowledge is prohibitive for large studies and routine clinical applications. Therefore these use cases require bioinformatics workflows to generate short lists of prioritized candidates. The germline analysis pipeline outlined here, by employing trio sequencing or larger pedigree sequencing data, narrows the field of candidates variants to identify causal variants for specific diseases. In this way, a shorter list of candidate variants can be generated for streamlining downstream analysis.
Generally, workflows for identification of disease-relevant variants rely on prefiltering steps to remove common polymorphisms and technical artifacts using publicly available and private databases. Of note, the majority of samples in public databases such as ExAC and 1000 Genomes originate from the Central European Population (CEP). Although the majority of samples analyzed in our study originated from the CEP, the combined MAF from all populations was used. However, it is recommended to use population-specific MAF if samples are collected from non-CEP populations. In addition, pooling MAF from samples sequenced on the same platform and processed within the same pipeline is recommended in order to remove technical artifacts arising specifically from the sequencing platform and pipeline used.
After the filtering steps, predictions regarding the functional impact of variants were used to narrow down the set of candidates. Many tools that predict the degree of deleteriousness of a variant are based on sequence conservation at the affected position [20]. However, individual tools are often not in a good agreement, justifying efforts to use a consensus set of results obtained using multiple tools or to use meta-tools that combine the results of multiple individual prediction algorithms [8,16]. For these reasons, our workflow relied on the CADD score [18], which integrates multiple levels of information including conservation and functional data, for variant prioritization.
A piece of information that complements the deleteriousness of a variant relates to the intolerance of an affected gene to new functional mutations. The intolerance or constraint score of a particular gene is calculated from the deviation between the observed number of functional mutations in a gene in large populations from the expected number based on the total amount of variation in this gene [21]. However, the gene intolerance score can also be misleading; for example, it predicts major cancer predisposition genes to be tolerant to new functional mutations [20]. Therefore, since some subregions of genes are much more intolerant than other subregions to functional mutations, a region-based or exon-based intolerance score could achieve higher resolution and therefore lead to better predictions. Furthermore, as noted earlier in the results section, the gene intolerance score performs poorly for AR variants. Therefore, a new gene intolerance score calculated solely from homozygous variants of the gene should be used to prioritize variants for an AR inheritance model.
Current studies to identify causal variants in genetic diseases usually only focus on exonic functional variants and investigate only SNVs and small indels. Consequently, for up to 60% of investigated cases no causal variants could be identified [3,4,22]. A possible reason for failure in these cases may be that those causal variants are focal copy number variants (CNVs), structural variants (SVs), or intronic variants affecting transcript splicing, which cannot comprehensively be identified from WES data and thus require WGS for full exploration. Alternatively, causal variants might affect nonprotein-coding regions of the genome. While these variants can be identified from WGS data, their interpretation and use for prediction of functional effects are much more challenging than for coding variants. Recently, multiple efforts have been made to produce more accurate tools to predict effect of non-coding variants in Mendelian diseases [23,24] so that in the future inclusion of non-coding variants into prioritization workflows should be possible.
5. Conclusion
The rapid decrease in sequencing costs has opened the door to the widespread application of genome sequencing in research as well as in clinical diagnostic settings. As a consequence, the resulting massive sequence data production has made data analysis and interpretation a daunting task, especially in clinical genomics settings where bioinformatics resources may be insufficient. In this study we developed an automated variant identification and prioritization pipeline for identification of causal variants of genetic disorders. By classification of variants and genes into distinct tiers, this pipeline makes it easier to obtain and focus on a small set of candidate variants to streamline downstream analysis. The pipeline will continuously be updated by adding new more accurate tools and scores to improve pipeline performance and will eventually also incorporate SVs, CNVs, and non-coding variants. This strategy should enable the characterization of causal variants underlying genetic disorders and facilitate diagnosis of such disorders in clinical settings.
Acknowledgments
We thank and the Genomics and Proteomics Core Facility at the German Cancer Research Center for their excellent technical support and expertise. We further like to acknowledge the data management group (DMG) in the Division of Theoretical Bioinformatics at the German Cancer Research Center for managing the sequence data.
Author Contributions
NP and MS developed the strategies and NP developed the pipeline. MS, CRB and RE provided the guidance and support. MG, CE and KH provided the samples and SW provided the sequencing data. NP, MS and RE wrote the manuscript. All the authors have read and contributed to the manuscript.
Funding
This work was supported by the BMBF-funded Heidelberg Center for Human Bioinformatics (HD-HuB) within the German Network for Bioinformatics Infrastructure (de.NBI) (#031A537C).
Competing Interests
Authors have no competing interests.
References
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010; 42: 30-35. [CrossRef] [Google scholar] [PubMed]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011; 12: 745-755. [CrossRef] [Google scholar] [PubMed]
- Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. Jama. 2014; 312: 1880-1887. [CrossRef] [Google scholar] [PubMed]
- Evers C, Staufner C, Granzow M, Paramasivam N, Hinderhofer K, Kaufmann L, et al. Impact of clinical exomes in neurodevelopmental and neurometabolic disorders. Mol Genet Metab. 2017; 121: 297-307. [CrossRef] [Google scholar] [PubMed]
- Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. New Engl J Med. 2014; 370: 2418-2425. [CrossRef] [Google scholar] [PubMed]
- Lelieveld SH, Veltman JA, Gilissen C. Novel bioinformatic developments for exome sequencing. Hum Genet. 2016; 135: 603-614. [CrossRef] [Google scholar] [PubMed]
- Consortium GP. A global reference for human genetic variation. Nature. 2015;526(7571):68-74. [CrossRef] [Google scholar] [PubMed]
- Pabinger S, Dander A, Fischer M, Snajder R, Sperk M, Efremova M, et al. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform. 2014; 15: 256-278. [CrossRef] [Google scholar] [PubMed]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009; 25: 1754-1760. [CrossRef] [Google scholar] [PubMed]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25: 2078-2079. [CrossRef] [Google scholar] [PubMed]
- Picard Tools - By Broad Institute [Internet]. [cited 2017 Aug 28]. Available from: http://broadinstitute.github.io/picard/index.html.
- Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SR, Wilkie AO, et al. Integrating mapping-, assembly-and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014; 46: 912-918. [CrossRef] [Google scholar] [PubMed]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22: 1760-1774. [CrossRef] [Google scholar] [PubMed]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38: e164-e164. [CrossRef] [Google scholar] [PubMed]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536: 285-291. [CrossRef] [Google scholar] [PubMed]
- Liu X, Jian X, Boerwinkle E. dbNSFP v2. 0: a database of human non‐synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013; 34: E2393-E2402. [CrossRef] [Google scholar] [PubMed]
- Cooper GM, Goode DL, Ng SB, Sidow A, Bamshad MJ, Shendure J, et al. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat Methods. 2010; 7: 250-251. [CrossRef] [Google scholar] [PubMed]
- Kircher M, Witten DM, Jain P, O'roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014; 46: 310-315. [CrossRef] [Google scholar] [PubMed]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine. 2015; 17: 405-423. [CrossRef] [Google scholar] [PubMed]
- Eilbeck K, Quinlan A, Yandell M. Settling the score: variant prioritization and Mendelian disease. Nat Rev Genet. 2017; 18: 599-612. [CrossRef] [Google scholar] [PubMed]
- Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013; 9: e1003709. [CrossRef] [Google scholar] [PubMed]
- Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016; 18: 696-704. [CrossRef] [Google scholar] [PubMed]
- Huang Y-F, Gulko B, Siepel A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat Genet. 2017; 49: 618-624. [CrossRef] [Google scholar] [PubMed]
- Smedley D, Schubach M, Jacobsen JO, Köhler S, Zemojtel T, Spielmann M, et al. A whole-genome analysis framework for effective identification of pathogenic regulatory variants in Mendelian disease. Am J Hum Genet. 2016; 99: 595-606. [CrossRef] [Google scholar] [PubMed]

