

Next Generation Sequencing and Pediatric Brain Tumors: Detection of Cancer Predisposition Syndromes in Patients and Their Families
Kerstin Grund 1,2,3 ![]() , Dominik Sturm 2,3,4
, Dominik Sturm 2,3,4![]() , Christian Sutter 1
, Christian Sutter 1![]() , Felix Sahm 5
, Felix Sahm 5![]() , Katrin Hinderhofer 1
, Katrin Hinderhofer 1![]() , Christian Kratz 6
, Christian Kratz 6![]() , Daniel Schrimpf 5
, Daniel Schrimpf 5![]() , Andreas von Deimling 5
, Andreas von Deimling 5![]() , Kristian W. Pajtler 2,3,4
, Kristian W. Pajtler 2,3,4![]() , David TW Jones 2,4
, David TW Jones 2,4![]() , Stefan M. Pfister 2,3,4
, Stefan M. Pfister 2,3,4![]() , Nicola Dikow 1,*
, Nicola Dikow 1,*![]()
- Institute for Human Genetics, University Hospital Heidelberg, Im Neuenheimer Feld 366, 69120 Heidelberg, Germany.
- Hopp-Children's Cancer Center at the NCT Heidelberg (KiTZ), Im Neuenheimer Feld 460, 69120 Heidelberg, Germany.
- Division of Pediatric Neuro-Oncology, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 280, 69120 Heidelberg, Germany, and German Cancer Consortium (DKTK), Heidelberg, Germany.
- Department of Pediatric Oncology, Hematology and Immunology, University Hospital Heidelberg, Im Neuenheimer Feld 430 , 69120 Heidelberg, Germany.
- Department of Neuropathology, Institute of Pathology, University Hospital Heidelberg, Im Neuenheimer Feld 224 and Clinical Cooperation Unit Neuropathology, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 224 69120 Heidelberg, Germany.
- Pediatric Hematology and Oncology, Hannover Medical School, Carl-Neuberg-Straße 1, 30625 Hannover, Germany.
* Correspondence: Nicola Dikow ![]()
Received: September 01, 2017 | Accepted: October 31, 2017 | Published: December 06, 2017
OBM Genetics 2017, Volume 1, Issue 4 doi:10.21926/obm.genet.1704011
Academic Editors: Ute Moog and Domenico Coviello
Recommended citation: Grund K, Sturm D, Sutter C, Sahm F, Hinderhofer K, Kratz C, Schrimpf D, Deimling AV, Pajtler KW, Jones DT, Pfister SM, Dikow N. Next Generation Sequencing and Pediatric Brain Tumors: Detection of Cancer Predisposition Syndromes in Patients and Their Families. OBM Genetics 2017;1(4):011; doi:10.21926/obm.genet.1704011.
© 2017 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
The study “Molecular Neuropathology 2.0” (MNP2.0) offers an integrated histo-molecular diagnosis including the detection of potential therapeutic targets for a large cohort of pediatric patients with primary CNS tumors. After obtaining parental and/or patient consent, in this study germline DNA analysis of all study subjects bridges the gap between scientific genetic analysis and medical care. The study’s workflow takes into consideration the conditions of a multicenter study, legal stipulations, as well as the need for close interdisciplinary cooperation. Here we present an elaborate workflow illustrated by four case studies of patients diagnosed with different cancer predisposition syndromes (CPS). The diagnosis of a CPS and subsequent family analysis are of substantial importance for all presented cases. Germline analysis within the ongoing MNP 2.0 study provides information about the prevalence and distribution of underlying germline mutations in a large population-based cohort of pediatric neuro-oncology patients. In addition, results of this study have the potential to identify high risk tumor entities- or molecular subgroups for underlying CPS.
Keywords
CPS; MNP2.0; study workflow; pediatric neuro-oncology; NGS panel sequencing; germline analysis
Background
The study “Molecular Neuropathology 2.0” (MNP2.0) provides molecular analyses for children and adolescents with primary brain tumors. Inclusion criteria include age younger than 21 years at the time of central nervous system (CNS) tumor primary diagnosis and availability of suitable tumor material for molecular analyses. Currently, the majority of all newly diagnosed pediatric CNS tumor patients are routinely registered for the MNP2.0 study in Germany. By analyzing DNA methylation patterns and performing targeted next-generation sequencing (NGS) on DNA from tumor tissue (and sequencing of blood for comparison), this study aims to improve CNS tumor classification and to identify possible targets for specific treatment agents.
Recent studies have shown a prevalence of cancer predisposition syndromes (CPS) as high as 5–8% in pediatric cancer patients [1,2,3]. Such rates are comparable to rates of underlying CPS in adult breast or colon cancer patients, where screening programs are well established [4,5]. The rate of CPS seems to be particularly high in the neuro-oncology subgroup of patients [2,3] based on a relatively low number of analyzed patients so far.
In addition to molecular analyses of tumor specimens, MNP2.0 includes analyses of DNA from peripheral blood cells in order to search for potential germline alterations. Such data provides information on CPS prevalence in unselected pediatric neuro-oncology patients, as well as the distribution of candidate disease-causing genes. Knowledge of the underlying germline mutation in a family may be relevant for various reasons. First, treatment decisions for the patient (e.g. treatment modalities, cumulative doses), surveillance programs for cancer and benign morbidities, or estimation of prognosis can depend on the underlying genetic alteration. Second, family members at risk can be identified and surveyed in appropriate programs (or released, if they have not inherited the pathogenic variant identified in their relatives). Finally, the knowledge of an underlying genetic mutation may affect quality of life (QoL) and stress levels of families seeking the cause of disease. However, despite these implications many features of patients with CPS still remain poorly understood, including (1) natural history/cancer epidemiology, (2) effectiveness of cancer surveillance, (3) genotype phenotype correlations, (4) toxicity of and response to standard cancer treatment protocols, (5) somatic mutation spectrum, and (5) optimal cancer treatment. Therefore, natural history studies, registries, and treatment protocols for patients with CPS are of high priority.
For CPS patients identified through MNP2.0, the disclosure of underlying germline mutations to patients and their families takes place at an interface between scientific analysis and a clinical diagnostic setting. Here, we present our concept on how to handle this interface in a multicenter and highly interdisciplinary environment in consideration of ethical guidelines and German legislation (Gendiagnostikgesetz, GenDG) regulating human genetic analyses. Practical procedures and consequences for patients and their families are illustrated using representative cases.
Ethics Statement
Molecular Neuropathology 2.0 is a prospective, multicenter study collecting clinical and molecular data with the goal to improve diagnostic accuracy for brain tumors in children. The study complies with the principles of the Declaration of Helsinki in its current version. Mandatory patient information and informed consent forms have been approved by the Ethics Committee of the University of Heidelberg.
Patients and Methods
Patients are referred to this study by the treating physician via the Brain Tumor Reference Center (Hirntumor-Referenzzentrum, HTRZ) at the Institute for Neuropathology, University Hospital Bonn, Germany. For each patient, a tumor tissue sample is accompanied by an EDTA-treated blood sample and both samples are shipped from Bonn to the Department of Neuropathology in Heidelberg (NP). In the consent form, the patients or the legal guardians state whether or not they wish to be informed about results of genetic germline analysis. Thus, participation in the MNP2.0 study is possible without receiving information about germline sequencing data.
DNA extraction from tumor tissue, sequencing, and bioinformatic analysis at NP has been described previously [6]. In parallel, DNA extraction from blood lymphocytes and NGS are performed to detect germline mutations. Sequence results of both sample types are compared to achieve more robust somatic mutation variant calling.
Out of 129 genes included in the panel of somatically sequenced genes, 45 are considered potentially relevant for patients and/or their families and were selected for germline analysis. The following criteria were applied for selection: (i) There must be published evidence for a clinical effect of germline variants in the selected gene. (ii) A causal connection of a germline mutation in the respective gene to the development of the respective tumor of the child must be known or conceivable, e.g. as the gene has been associated with oncogenesis or tumor predisposition. (iii) Knowledge about a mutation in the selected gene must offer medically actionable consequences for patients and/or their families. The selection criteria are summarized in Table 1 and a gene must meet all three mandatory criteria to be considered a candidate. The full list of selected genes is shown in Table S1.
Table 1 Criteria for selection of genes for germline analysis
|
|
|
Pre-filtered variant datasets from both tumor tissue and blood lymphocytes were sent to our accredited diagnostic molecular genetic laboratory within the Institute of Human Genetics (HG) for further assessment. Using Alamut Visual v.2.9/2.10 (Interactive Biosoftware, Rouen, France), variant information was drawn from international databases containing germline mutations associated with human genetic diseases and somatic mutations associated with cancer (ClinVar, LOVD 3.0, LOVD-Zhejiang (PR China site) HGMD professional, UMD, BIC, ARUP, the German Consortium for Hereditary Breast and Ovarian Cancer database, TCGA, COSMIC). In addition, in silico protein variant and splice prediction tools (Align GVGD, SIFT, PolyPhen-2, Mutation Taster, NNSPLICE, Human Splicing Finder, MaxEntScan, GeneSplicer, Human SpliceSiteFinder-like) were applied and information about variant frequencies in the general population were analyzed. For variant assessment, literature with regard to the functional impairment of gene variants or associations with respective genetic diseases was extracted from PubMed, OMIM, Google, and GoogleScholar and taken into account. Variants were classified according to the International Agency for Research on Cancer (IARC) classification criteria (class-1 “not pathogenic” through class-5 “definitively pathogenic” [7]). Classes 1–2 are defined as “benign,” classes 4–5 as “pathogenic,” and class-3 as variants of unknown significance (VUS). Only variants of classes 4–5 are reported back to the NGS laboratory. Aliquots (0.5-1.0 µg each) of blood leukocyte DNA with suspicious NGS findings were sent to the diagnostic molecular genetic laboratory of the Institute of Human Genetics at Heidelberg for validation by Sanger sequencing.
For validation of suspicious sequence variants identified by NGS screening, 50–100 ng of genomic leukocyte DNA were amplified by polymerase chain reaction (PCR) with primers with M13 (forward) or p172 (reverse) universal adapters for respective exons. Subsequent DNA sequencing was performed using M13 (forward) or p172 (reverse) primers. Electrophoresis of the sequence products was performed using a 3130xl Genetic Analyzer (Applied Biosystems/Life Technologies/ThermoFisher, Darmstadt, Germany). Electropherograms were analyzed using the SeqPilot software (v.4.2.2, JSI-Medisys, Ettenheim, Germany).
Results
Workflow
The following workflow has been established for patients with pediatric CNS tumors enrolled in the MNP2.0 study and is depicted in Figure 1. Written parental/patient consent and patient choice regarding disclosure of the results of germline analysis are included. Patients are enrolled by the treating physician via the Brain Tumor Reference Center (Hirntumor-Referenzzentrum, HTRZ) at the Institute for Neuropathology, University Hospital Bonn.
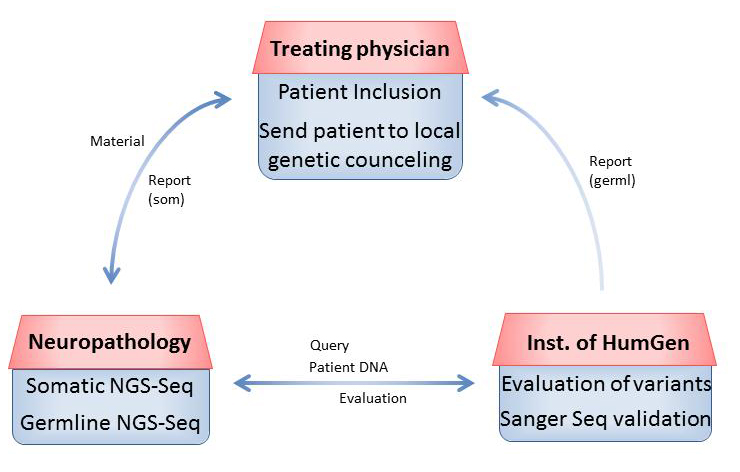
Figure 1 Workflow of germline diagnostics within MNP2.0 in a schematic overview.
Tumor material and a matching blood sample of the patient are then shipped to the Department of Neuropathology, University of Heidelberg (NP). DNA is extracted from both tumor and blood. Targeted NGS is next performed for both sample types using a gene panel of 129 genes known to be involved in brain tumorigenesis or frequently mutated in brain tumor tissue [6].
Germline variants with potential significance among the 45 of 129 genes selected for analysis are sent to the Institute of Human Genetics, Heidelberg University (HG) together with available clinical information for evaluation. Only variants possibly responsible for the patient’s disease and classified as “likely” pathogenic (classes 4–5) are subjected to further confirmation by Sanger sequencing by the diagnostic molecular genetic laboratory (HG).
The treating physician receives a written report by NP regarding the DNA methylation profile of the tumor and the somatic mutations including a remark that a.) no germline variants were detected that are currently regarded as relevant or b.) a DNA sample has been sent to the molecular genetic laboratory (HG) for validation and interpretation of potential germline variants.
(“Likely” pathogenic variants are confirmed in the molecular genetic laboratory (HG) and reported to the treating physician accompanied by a brief summary of associated risks and clinical consequences. A comment referring to the German Gendiagnostikgesetz (GenDG) expresses that genetic counseling must be offered to the patient or his/her parents. Genetic counseling is always offered by HG in Heidelberg, but alternatively may take place at the local institution of human genetics. Standard procedures include dispensing of information about the variant and the gene, associated risks, clinical recommendations for the patient and individuals at risk, and the possibility of predictive testing of family members.
For the assessment, validation, and discussion of germline variants detected within the MNP2.0 study, experts from the fields of pediatric oncology, neuropathology, and human genetics are in close contact. A monthly interdisciplinary board has been established to discuss all cases with a suspected germline component individually, as well as to address any methodological questions.
Case reports
Clinical and molecular data of four representative patients are summarized in Table 2.
Patient 1 is a girl diagnosed with a medulloblastoma at the age of 2 years. Germline panel analysis revealed a heterozygous SUFU variant (c.455-1G>A (HGVS) in Intron 3). SUFU sequence variants have been associated with autosomal dominant nevoid basal cell carcinoma syndrome (NBCCS, Gorlin syndrome). Young patients are at an increased risk for medulloblastoma (33% [8]) and, beginning at puberty, other tumors, such as numerous basal cell carcinomas. For affected patients and healthy family members at risk, surveillance programs have been established [9]. From childhood onwards, direct sunlight should be avoided and the skin should be protected by clothing or sunblock. If radiotherapy is required, special skin-sparing precautions should be taken in order to avoid massive development of basal cell carcinomas in the respective area. After genetic counseling, predictive testing of the parents and the younger brother of patient 1 revealed no SUFU germline mutations; therefore no specific surveillance was required for them. Recurrence risk for additional children of the couple is low (< 1%, due to potential rare germline mosaicism). Recurrence risk for future children of the young patient is 50%.
Patient 2 is a 9-year-old boy with a CNS melanoma diagnosed at the age of 9 years. Concurrently, this patient visited the genetic outpatient clinic for evaluation of obvious neurocutaneous melanosis (NCM). Consistent with the assumption that NCM is a non-inherited disease, germline analysis of leukocyte DNA revealed no detectable mutations, while analysis of tumor tissue and of a cutaneous nevus biopsy revealed a common heterozygous NRAS variant c.181C>A (HGVS), p.(Gln61Lys). No loss of heterozygosity was detected in tumor DNA, but an additionally acquired mutation in the CDKN2A/B gene was found that is associated with the development of malignant melanoma [10]. As patient 2 carried the NRAS-mutation only as a postzygotic mosaicism, inheritance from his parents was very unlikely and no family members were tested. Examination of his parents and two brothers revealed no cutaneous signs of a neurocutaneous melanosis (giant cell nevi). The patient himself is at increased risk for melanoma both in the CNS and skin, requiring a surveillance program [11]. However, he is in palliative care as complete resection of the CNS melanoma is impossible.
Patient 3 is a 12-year-old girl with a pilocytic astrocytoma diagnosed at the age of 11 years. Germline analysis revealed a heterozygous variant in MSH6 c.219C>T (HGVS), p.(Arg1035*). MSH6-mutations are associated with the autosomal dominant and well-known cancer predisposition syndrome Lynch-syndrome). Patients are at an increased risk for colorectal cancer, endometrial cancer, and at a lower risk for other cancers including ovary, small bowel, gastric, urinary tract, skin, hepatobiliary tract, and brain [5]. The occurrence of brain tumors in families with colorectal cancer predisposition syndromes was historically termed “Turcot’s syndrome.” Beneficial surveillance programs starting at the age of 25 years have been well established [5]. During genetic counseling, the family history was assessed and showed maternal family members with colorectal cancer and one third-degree relative who died in early childhood from a brain tumor. Predictive testing of the parents of patient 3 identified her mother as a healthy carrier of the pathogenic MSH6-variant. The standardized surveillance for gastrointestinal and endometrial cancer has been initiated for the mother and genetic counseling has been offered to her relatives. The patient herself is not in complete remission, although she carries a very slow growing tumor. The surveillance program for her will start at the age of 25 [12].
Patient 4 is a 12-year-old boy diagnosed with a glioblastoma (GBM) at the age of 11 and acute lymphoblastic leukemia (ALL) at the age of 2 years. A heterozygous mutation in ATM (c.6199-1G>T (HGVS)) was identified in this patient. Biallelic ATM variants have been associated with ataxia telangiectasia, a CPS with a high risk for malignancies that are predominantly leukemias and lymphomas, and other features such as gait and truncal ataxia in early childhood [13]. In contrast, heterozygous ATM variants are mainly associated with an increased risk of breast cancer for females and therefore surveillance recommendations have been established [14,15]. Family history documented during genetic counseling showed parental consanguinity and breast cancer in the father’s mother. As ALL and GBM in the young patient 4 are not sufficiently explained by a heterozygous ATM mutation, NGS data were reevaluated for ATM variants, but no further variant was detected, including class 3 variants. Predictive testing of the parents showed paternal inheritance of the patient’s ATM-variant. Although no surveillance program exists for male heterozygous mutation carriers, predictive testing has been offered to his female relatives.
Table 2 summary of patient data
|
|
Patient 1 |
Patient 2 |
Patient 3 |
Patient 4 |
|
Age at diagnosis |
2y |
9y |
11y |
11y |
|
Sex |
f |
m |
f |
m |
|
Tumor |
Medulloblastoma |
CNS melanoma |
Astrocytoma |
Glioblastoma |
|
Additional disease |
|
Neurocutaneous melanosis |
|
ALL at age 2y |
|
Mutated gene |
SUFU |
NRAS (mos.) |
MSH6 |
ATM |
|
Mutation |
c.455-1G>A
|
c.181C>A, p.(Gln61Lys) |
c.3103C>T, p.(Arg1035*) |
c.6199-1G>T |
|
Inheritance |
het. de novo |
postzygot. mosaicism |
het. (maternal) |
het. (paternal) |
|
Healthy mutations carriers (HMC) in family |
no |
no |
yes |
yes |
|
Surveillance recommendations |
yes, patient |
yes, patient |
yes, patient and mother |
no * |
|
Status of patient |
Complete remission |
Palliative care |
Progressive disease |
Stable disease |
| * Only for female mutation carriers in paternal family | ||||
Discussion
In Germany, the “Gendiagnostikgesetz” (GenDG; Gene Diagnostics Law) provides the legal framework for genetic testing procedures as well as for predictive testing (presymptomatic testing of healthy individuals) in a diagnostic setting of patients affected by a presumably genetic disease. Prior to genetic analysis, patients must be informed about analyzed genes and consequences for themselves and their relatives. In contrast, genetic analyses in scientific studies are not subject to the GenDG, but written informed consent consistent with a study protocol approved by institutional ethics committees must be given. Germline analysis within the MNP2.0 study enables detection of CPS-associated germline mutations in a large population-based cohort of children with CNS tumors. The disclosure of results of germline analysis takes place at an interface between research and clinical settings, especially when the genetic information affects family members that are not subjects of the study. Therefore, the patients or their parents must decide prior to registration for the MNP2.0 study whether germline mutations in “relevant genes related to the disease” should be divulged to them or not. We assessed a list of “selection criteria” (Table 1) and defined “relevant” genes. The criteria mirror questions frequently mentioned by parents during genetic counseling: “Why did the tumor occur in our child?”, “Are there any practical consequences regarding the treatment of the affected child or surveillance options?", and "Is the brother/sister/other family member at risk for the same or other tumor?” On the one hand, this selection of genes may reduce the sensitivity of CPS detection. On the other hand, the selection prevents disclosure of variants in genes that cannot be interpreted in the context of the patient's individual and family history, and where a patient’s subjective uncertainty would negate any benefit.
A genetic diagnosis of CPS may have practical and psychological aspects. For all patients presented here, the diagnosis of CPS reveals practical consequences for patients and/or their families. Consequences include recommendation of surveillance programs for the patient or healthy mutation carriers or the release of family members not carrying the disease-causing variant. Depending on the affected gene, family members at risk can be included in prevention and surveillance programs, as illustrated for patient 3. The benefit of such programs has been established for common CPS such as familial breast and ovarian cancer [4], hereditary colorectal cancer [5], neurofibromatosis [16,17], or multiple endocrine neoplasia [18,19]. Also, for very rare CPS such as DICER1 syndrome or hereditary paraganglioma/pheochromocytoma syndrome, surveillance recommendations exist [20,21,22] and recent data show advantages for patients with TP53 variants under screening protocols [23,24]. A benefit of screening for rare pediatric CPS is assumed but needs to be evaluated in ongoing studies [25]. Moreover, CPS may also impact treatment decisions (e.g. radiation versus chemotherapy) or prognostic implications.
Whether germline analysis is psychologically beneficial for patients or mainly induces additional stress is controversial [26]. Parents of patients presented here reported significant psychological burden due to the nature of their child’s malignant disease and treatment. Moreover, they were not informed about the detected germline variant prior to genetic counseling (HG). However, they were all interested in the underlying cause of their child’s tumor, information on treatment, and surveillance options for the young patient, siblings, or other relatives, and decided to receive predictive testing of relatives. The parents of the four patients perceived no need for specialized psychological consultation; one family mentioned that in the course of the child’s disease they “got used to bad news.” Therefore, finding a germline mutation as the underlying cause was “minor” compared with the disclosure of the oncologic disease itself. In families with children with malformation syndromes or intellectual disabilities, knowledge of an underlying genetic cause has been associated with better quality of life (QoL) and lower psychological burden for the parents [27,28]. Important factors may be the perceived alleviation of guilt, as well as the reestablishment of control through increased understanding of prognosis, management, risk of recurrence for family members, or family planning. Nevertheless, immediately following disclosure of a CPS, the family may indeed suffer from additional stress, but long-term impact on QoL or psychological burden of such families has not yet been evaluated. Meanwhile, long-term follow up of families affected by hereditary breast and ovarian cancer revealed no evidence of mental distress caused by the knowledge of an underlying germline mutation [29,30].
The MNP2.0 study workflow outlined here contains a number of limitations with regard to diagnostic sensitivity. First, the family history is unknown to the researchers. In the clinical diagnostic setting, the spectrum of tumors in the family may allow a clinical diagnosis (such as Li-Fraumeni syndrome [31,32,33,34]) or point to a specific group of genes. A VUS in the respective gene might be assessed differently and after segregation analysis and/or functional testing be reevaluated as a probable cause of disease. In contrast, a VUS would not be reported in the present study. In a similar way, detailed knowledge of the clinical phenotype evaluated by experienced clinical geneticists would enhance the sensitivity for the detection of CPS, for example if the phenotype points to phacomatoses such as tuberous sclerosis complex or to syndromes associated with dysmorphic features and an elevated tumor risk (e.g., RASopathies such as Noonan syndrome).
With the current workflow, patient 2 would not have been identified as a CPS patient even though he harbored a postzygotic mosaic variant of NRAS, since his blood DNA did not carry any detectable pathogenic mutations. A growing number of cancer predispositions linked to postzygotic mosaicism might cause a comparably high individual risk as established in familial CPS [11]. However, these patients usually present with a recognizable phenotype. Due to appreciation of a pathognomonic phenotype, patient 2 was referred for genetic assessment and counseling. Additional patients might be included in MNP2.0 that were not identified as CPS patients for the same reason. Therefore, knowledge of the patient’s phenotype is crucial for the detection of a CPS caused by postzygotic mosaicism.
For MNP2.0, in order to enhance diagnostic sensitivity (e.g. with respect to handling of VUS or for the detection of postzygotic mosaicism) additional questionnaires will be provided for new patients [35]. These questionnaires will serve to collect additional clues to detect an underlying CPS, such as phenotypic abnormalities or a personal or family history of cancer. Moreover, the implementation of an interdisciplinary consultation, in which pediatric neuro-oncology patients are seen by a clinical geneticist prior to molecular genetic analysis, is expected to further increase sensitivity and specificity of CPS detection. However, the feasibility of such an approach used nationwide is unclear.
From this ongoing study we also expect answers to the following questions: How many of the families with detected germline variants seek genetic counseling or decide to submit to predictive testing? Are there any other hints in the families pointing to CPS? How many patients with CPS have de novo mutations? The statistical evaluation of the frequency of CPS in pediatric neuro-oncology patients, frequently affected genes, or tumor entities with markedly increased risk for CPS is still pending. Likewise, many open questions remain regarding the clinical/biologic features of individuals with various CPS (natural history, genotype-phenotype correlations, genetic modifiers, somatic mutation spectrum, treatment response, and toxicity, etc.) and collection of data through international registries is urgently needed.
Conclusions
The interdisciplinary approach presented here provides tumor and germline NGS and clinical care of affected patients and their families with CPS. Such approaches are increasingly important in settings where diagnostic tools are emerging at the interface between scientific analysis and clinical genetics. The sensitivity of detection of CPS is expected to be enhanced with collection of more information on the patient’s phenotype and family history prior to NGS analysis. In this study, for families with children suffering from CNS tumors, knowledge of the underlying CPS had various relevant practical consequences.
Acknowledgement
All authors contributed to the data collection and analysis and the writing of this review.
Competing Interests
The authors have declared that no competing interests exist.
References
- Narod SA, Stiller C, Lenoir GM. An estimate of the heritable fraction of childhood cancer. Brit J Cancer. 1991;63(6):993-999. [CrossRef]
- Ripperger T, Bielack SS, Borkhardt A, Brecht IB, Burkhardt B, Calaminus G, et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am J Med Genet Part A. 2017;173(4):1017-1037. [CrossRef]
- Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. New Engl J Med. 2015;373(24):2336-2346. [CrossRef]
- Couch FJ, Nathanson KL, Offit K. Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science. 2014;343(6178):1466-1470. [CrossRef]
- Lynch HT, de la Chapelle A. Hereditary colorectal cancer. New Engl J Med. 2003;348(10):919-932. [CrossRef]
- Sahm F, Schrimpf D, Jones DT, Meyer J, Kratz A, Reuss D, et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta neuropathol. 2016;131(6):903-910. [CrossRef]
- Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29(11):1282-1291. [CrossRef]
- Smith MJ, Beetz C, Williams SG, Bhaskar SS, O'Sullivan J, Anderson B, et al. Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol: official journal of the American Society of Clinical Oncology. 2014;32(36):4155-4161. [CrossRef]
- Foulkes WD, Kamihara J, Evans DGR, Brugieres L, Bourdeaut F, Molenaar JJ, et al. Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin Cancer Res: an official journal of the American Association for Cancer Research. 2017;23(12):e62-e67.[AACR]
- Soura E, Eliades PJ, Shannon K, Stratigos AJ, Tsao H. Hereditary melanoma: Update on syndromes and management: Genetics of familial atypical multiple mole melanoma syndrome. J Am Acad Dermatol. 2016;74(3):395-407; quiz 408-310.[ScienceDirect]
- Kinsler VA, Thomas AC, Ishida M, Bulstrode NW, Loughlin S, Hing S, et al. Multiple congenital melanocytic nevi and neurocutaneous melanosis are caused by postzygotic mutations in codon 61 of NRAS. J Invest Dermatol. 2013;133(9):2229-2236. [CrossRef]
- Pox C, Aretz S, Bischoff SC, Graeven U, Hass M, Heussner P, et al. [S3-guideline colorectal cancer version 1.0]. Z Gastroenterol. 2013;51(8):753-854. [CrossRef]
- Taylor AM, Lam Z, Last JI, Byrd PJ. Ataxia telangiectasia: more variation at clinical and cellular levels. Clin Genet. 2015;87(3):199-208. [CrossRef]
- Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017. [CrossRef]
- Southey MC, Goldgar DE, Winqvist R, Pylkas K, Couch F, Tischkowitz M, et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet. 2016;53(12):800-811. [CrossRef]
- Evans DGR, Salvador H, Chang VY, Erez A, Voss SD, Druker H, et al. Cancer and Central Nervous System Tumor Surveillance in Pediatric Neurofibromatosis 2 and Related Disorders. Clin Cancer Res: an official journal of the American Association for Cancer Research. 2017;23(12):e54-e61.[AACR]
- Evans DGR, Salvador H, Chang VY, Erez A, Voss SD, Schneider KW, et al. Cancer and Central Nervous System Tumor Surveillance in Pediatric Neurofibromatosis 1. Clin Cancer Res: an official journal of the American Association for Cancer Research. 2017;23(12):e46-e53.[AACR]
- Machens A, Dralle H. Therapeutic Effectiveness of Screening for Multiple Endocrine Neoplasia Type 2A. J Clin Endocr Metab. 2015;100(7):2539-2545. [CrossRef]
- Wasserman JD, Tomlinson GE, Druker H, Kamihara J, Kohlmann WK, Kratz CP, et al. Multiple Endocrine Neoplasia and Hyperparathyroid-Jaw Tumor Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res: an official journal of the American Association for Cancer Research. 2017;23(13):e123-e132.[AACR]
- Doros L, Schultz KA, Stewart DR, Bauer AJ, Williams G, Rossi CT, et al. DICER1-Related Disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. Gene Rev (R). Seattle (WA)1993.[AACR]
- Rednam SP, Erez A, Druker H, Janeway KA, Kamihara J, Kohlmann WK, et al. Von Hippel-Lindau and Hereditary Pheochromocytoma/Paraganglioma Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res: an official journal of the American Association for Cancer Research. 2017;23(12):e68-e75.[AACR]
- Schultz KAP, Rednam SP, Kamihara J, Doros L, Achatz MI, Wasserman JD, et al. PTEN, DICER1, FH, and Their Associated Tumor Susceptibility Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res: an official journal of the American Association for Cancer Research. 2017;23(12):e76-e82.[AACR]
- Villani A, Shore A, Wasserman JD, Stephens D, Kim RH, Druker H, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol. 2016;17(9):1295-1305. [CrossRef]
- Kratz CP, Achatz MI, Brugieres L, Frebourg T, Garber JE, Greer MC, et al. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin Cancer Res: an official journal of the American Association for Cancer Research. 2017;23(11):e38-e45.[AACR]
- Brodeur GM, Nichols KE, Plon SE, Schiffman JD, Malkin D. Pediatric Cancer Predisposition and Surveillance: An Overview, and a Tribute to Alfred G. Knudson Jr. Clin Cancer Res: an official journal of the American Association for Cancer Research. 2017;23(11):e1-e5.[AACR]
- Druker H, Zelley K, McGee RB, Scollon SR, Kohlmann WK, Schneider KA, et al. Genetic Counselor Recommendations for Cancer Predisposition Evaluation and Surveillance in the Pediatric Oncology Patient. Clin Cancer Res: an official journal of the American Association for Cancer Research. 2017;23(13):e91-e97.[AACR]
- Lenhard W, Breitenbach E, Ebert H, Schindelhauer-Deutscher HJ, Henn W. Psychological benefit of diagnostic certainty for mothers of children with disabilities: lessons from Down syndrome. Am J Med Genet Part A. 2005;133A(2):170-175. [CrossRef]
- Lingen M, Albers L, Borchers M, Haass S, Gartner J, Schroder S, et al. Obtaining a genetic diagnosis in a child with disability: impact on parental quality of life. Clin Genet. 2016;89(2):258-266. [CrossRef]
- Michelsen TM, Dorum A, Dahl AA. A controlled study of mental distress and somatic complaints after risk-reducing salpingo-oophorectomy in women at risk for hereditary breast ovarian cancer. Gynecol Oncol. 2009;113(1):128-133. [CrossRef]
- Oberguggenberger A, Sztankay M, Morscher RJ, Sperner-Unterweger B, Weber I, Hubalek M, et al. Psychosocial outcomes and counselee satisfaction following genetic counseling for hereditary breast and ovarian cancer: A patient-reported outcome study. J Psychosom Res. 2016;89:39-45. [CrossRef]
- Chompret A, Abel A, Stoppa-Lyonnet D, Brugieres L, Pages S, Feunteun J, et al. Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet. 2001;38(1):43-47. [CrossRef]
- Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ, et al. Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol: official journal of the American Society of Clinical Oncology. 2009;27(8):1250-1256. [CrossRef]
- Ruijs MW, Verhoef S, Rookus MA, Pruntel R, van der Hout AH, Hogervorst FB, et al. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet. 2010;47(6):421-428. [CrossRef]
- Tinat J, Bougeard G, Baert-Desurmont S, Vasseur S, Martin C, Bouvignies E, et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol: official journal of the American Society of Clinical Oncology. 2009;27(26):e108-109; author reply e110.[AACR]
- Jongmans MC, Loeffen JL, Waanders E, Hoogerbrugge PM, Ligtenberg MJ, Kuiper RP, et al. Recognition of genetic predisposition in pediatric cancer patients: An easy-to-use selection tool. Eur J Med Genet. 2016;59(3):116-125. [CrossRef]
